The package contains (once unzipped by Winzip) :
nochaos.exe the executable (32 bits for Windows 95 and possibly
NT - not for 16 bits Win 3 or DOS).
nochaos.dat a test file.
Ask for the Fortran 77 or
C sources if needed (was compiled by f2c and MSVC++2.0).
Configuration files (.cfg) for a Reverse Monte Carlo modelling are usually built up in two ways. A random configuration is created by the RANDOM software and then the atoms are moved to satisfy cut-off restrictions by the MOVEOUT program, this may be quite long. The second way is to create a crystal configuration by using the CRYSTAL program. A third possibility is here offered by NOCHAOS which fills the box progressively with random positions only if the cut-off distances are satisfied. The process is much faster than the RANDOM/MOVEOUT combination. Nevertheless, the user should first enter the atoms which are expected to be at the largest distances from themselves and those at the smallest distances from themselves at the end. Moreover, if the cut-off are selected too large, then finding places for the later atoms may become quite long, if not impossible. In case of impossibility to fill the box in a resonable time (usually a few minutes on a Pentium II 266 MHz), reduce the cut-off values.
Getting Started
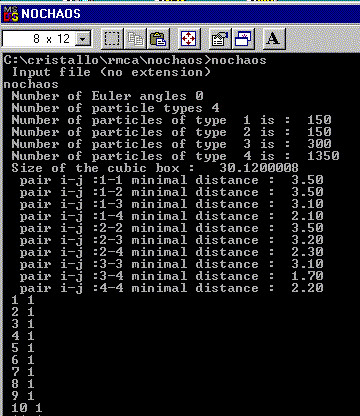
The NOCHAOS program for PC starts by a double click on its name in the Windows Explorer (or typing 'nochaos' in a DOS box opened in the directory containing nochaos.exe and the data files). A window is opened. You are prompted to give the entry filename (here nochaos, the test file) :
The input data are first listed on the screen. Then the filling
of the box starts by the first atom-type, which should stand at the largest
distances from the same atom-type neighbours (and then the second, etc,
up to the last atom showing the smallest distances with its same atom-type
neighbours). Usually, for ionic compounds, the cations with the largest
ionic radii should be the first atoms to be entered in the box (Na, Pb
before Fe in the test file). The anions will be the last (F in the test
file). Each time an atom is entered in the box, its number together with
its number-atom-type are given on the screen, allowing to observe if there
is some difficulty at placing some atoms (the speed decreases). In case
of real difficulty, the user should lower the appropriate cut-off value(s).
At the end, a configuration file is created, ready for applying the RMCA
software.
For more info, get the manual RMCA.ps and the Fortran source code at
the RMCA FTP server.
Windows 95 versions of RMCA, RANDOM, MOVEOUT and CRYSTAL are also available