STEP 4B
How to examine the 'Solution' using .RES
and .INS files
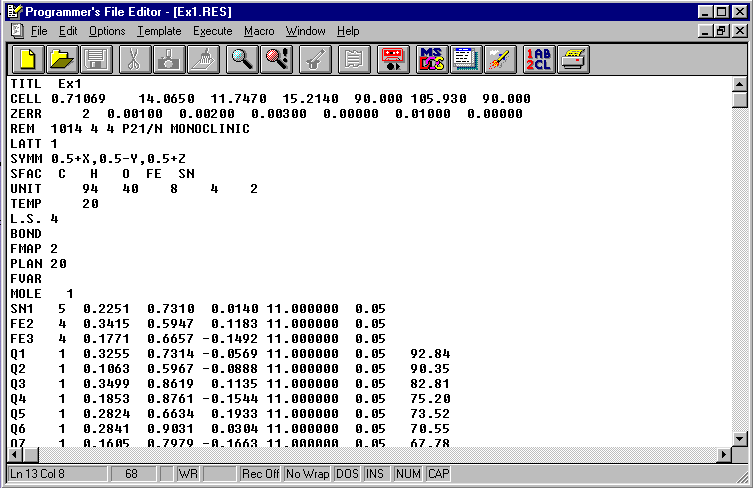
At this point, it is normal to delete rubbish and to start to refine 'good' atoms
The heavy atoms Sn1, Fe2
and Fe3 can be assumed good and all else rubbish.
These atoms should be enough to phase the map.
Select Edit.RES (SHELXS-97
results file).
Increase the PLAN to PLAN 40
Put HKLF 4 after FE3
The MOLE instruction is not required.
Save the now edited .RES file as a .INS file (instruction file) for SHELXL-97/2 imput.
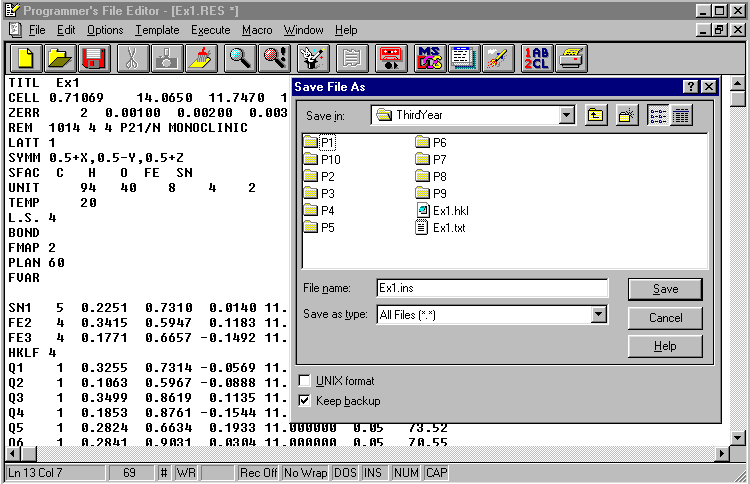
Select Run Job ![]() and SHELXL-97/2.
and SHELXL-97/2.
Notice that the wR2 is dropping through the cycles.
answer Yes to Overwrite .INS ?
Select Edit.INS
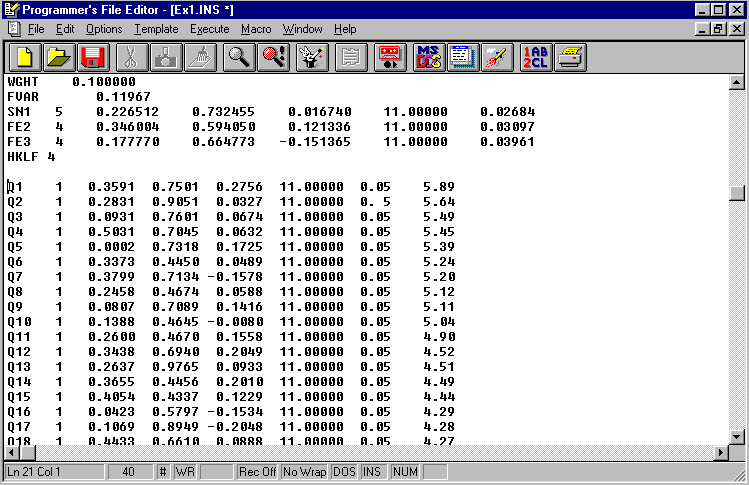
The refined Sn and Fe(s) have reasonable thermal parameters a good value is close to 0.04.
The peaks in the difference map have been given
the atom type number of carbon (1) and the
label Q.
Notice the sharp cut-off after Q32.
Add Qs from the Difference
Map using copy and paste.
In this case peaks Q1 to Q32 are pasted just above the HKLF 4
instruction.
Save the file and exit PFE.
Select Run Job ![]() and SHELXL-97/2.
and SHELXL-97/2.
The wR2 value will now drop further.
answer Yes to Overwrite .INS ?
Select Edit.INS
The thermal parameter is in the last column and the 'atoms' with parameters greater
than 0.10 are probably just rubbish. Delete these and put ANIS 3
before
Sn1. ANIS 3 will allow the three metal atoms to refine anisotropically (as ellipsoids).
This will remove 'ghost peaks' which tend to appear around heavy atoms when refined
isotropically (as spheres).
Save the file and exit PFE.
Select Run Job ![]() and SHELXL-97/2
and SHELXL-97/2
The wR2 value will now drop further.
answer Yes to Overwrite .INS ?
Select Run Job ![]() and ORTEX (default)
and ORTEX (default)
Answer Yes to Use Defaults?
Use .INS (default) as imput source.
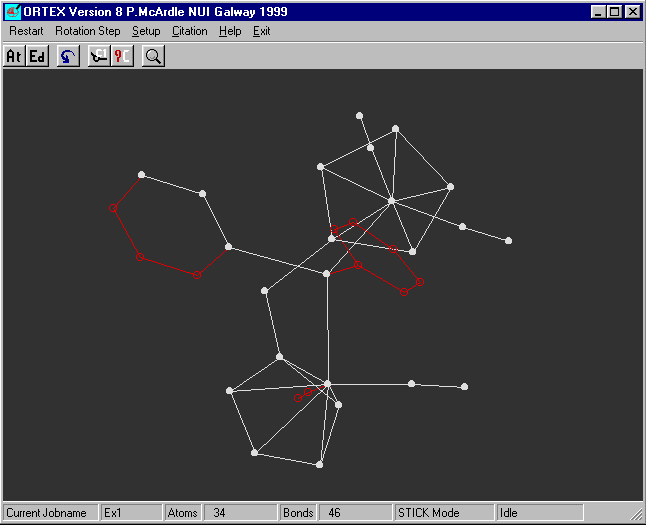
The arrow keys are used to rotate the molecule
for y Rotation use the left/right cursor keys, the up/down keys for z
and shift + left/right keys for x
If you rotate the molecule you will see that all looks reasonable.
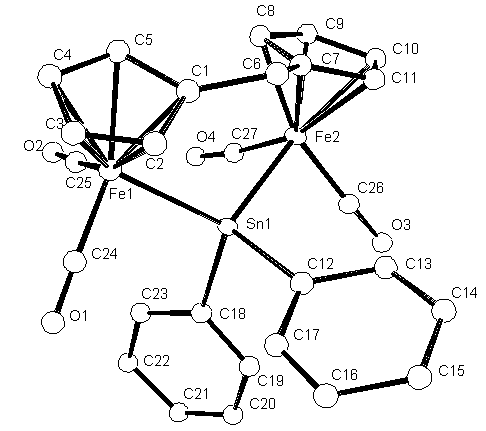
It is now necessary to name all the atoms with reasonable names.
Number the Carbons from C1 up to C27 and the four Oxygens from O1 - O4.
The oxygens are on the CO groups attached to the Fe atoms.
To rename the atoms select Edit ![]() on the
ORTEX (stick) Menu.
on the
ORTEX (stick) Menu.
You need to be careful. In Edit atoms are selected with the mouse
and the atom's name is shown on the bottom left. When an atom is
selected three actions are possible.
1. Click a blank area of the screen to clear the selector.
2. Press ![]() (or C on the keyboard) and you can change an
atoms name.
(or C on the keyboard) and you can change an
atoms name.
3. Press ![]() (or D on keyboard) to delete an atom.
(or D on keyboard) to delete an atom.
Change (rename) all of the Carbon and Oxygen atoms and then
Select Update (this will
overwrite the old .INS file)
Select the Default options.
The result of the ORTEX operations are now written to EX1.INS
The Carbon atoms are last and sorted in numerical order.
Before this file is sent to SHELXL-97/2 use Edit.INS to
check that all atoms have Unique Names. Duplicate names will
cause SHELXL to fail. If you have made a mistake and have 2 C13s
say then just change one of them to C53 for the moment.
Select Run Job ![]() and SHELXL-97/2.
and SHELXL-97/2.
The wR2 factor will now decrease again. If the maximum and minimum
peaks in the Difference map are not greater than 1.0 (absolute value) then
you have found and refined all of the non-H atoms.
To add hydrogen atoms the SHELX HFIX command can be used.
Use Edit.INS and put in the following three lines after
FMAP 2
HFIX 43 -1.3 C2 C3 C4 C5 C8 C9 C10 C11
HFIX 23 -1.3 C6
ANIS
(where 43 is for sp2 type CH and 23 is for secondary CH2 the -1.3
asks for the H to have a thermal paramater 30% less than its C and ANIS
will allow all non-H to refine anisotropically)
Select Run Job ![]() and SHELXL-97/2
and SHELXL-97/2
Answer Yes to Overwrite .INS ?
EX1 should now be fully refined.