PRODD
Profile Refinement of Diffraction Data
using the
Cambridge Crystallographic Subroutine Library
(CCSL)
J P Wright & J B Forsyth
Rutherford Appleton Laboratory Report RAL-TR-2000-012
Version 1.0
May 2000
This report describes extensions to the profile refinement code first developed at the Rutherford Appleton Laboratory in the 1980s to treat time-of-flight powder neutron diffraction data collected at ISIS. The bulk of the earlier design and coding was carried out by W I F David and the late J C Matthewman, who chose to augment the basic subroutines for crystallographic calculations contained in the Cambridge Crystallographic Subroutine Library (CCSL)[1] by adding a new Profile Refinement (PR) library of subroutines, a library of Peak shape Functions (PF) and an associated set of Profile MAin programs (PMA) [2].
Early versions of these main programs (TF12LS, CN13LS, SR15LS etc) were limited to the treatment of diffraction patterns from a single-phase, non-magnetic sample. Moreover, they could only be used to analyse one type of data at a time, eg time of flight (ToF) neutron, constant wavelength neutron or X-ray data. Code for multiphase analysis was added by the same authors in the early 1990’s and many of the PR Library subroutines were modified in 1995-1997 by P J Brown & J B Forsyth to include neutron magnetic scattering from a single phase sample.
The present authors have now extended the CCSL code for profile refinement in two directions. Firstly, it may be used to fit neutron data from a multiphase sample in which some or all of the phases are magnetically ordered. Secondly, but perhaps equally importantly, data in more than one histogram of intensity versus angle or time-of-flight may be treated within a single refinement. The latter development is crucial to diffraction at pulsed neutron sources, where a powder diffractometer is usually equipped with a number of detector banks having significantly different resolution characteristics. Although the latter development was foreseen by earlier contributors to the profile refinement code [2], the premature death of Judy Matthewman in 1993 delayed further progress in this area.
We should also like to acknowledge the careful work carried out by K Shankland who subjected a 1995 version of the basic CCSL library to several different Fortran compilers running on a variety of computers. This exercise revealed a number of infelicities in the code which were removed. Shankland was also responsible for demonstrating the method of running the profile refinement code, and indeed any CCSL program, on a personal computer using G77 Fortran.
2. PRODD
We have chosen to name the new computer program PRODD, which is an acronym for Profile Refinement of Diffraction Data. PRODD allows one to refine data from samples containing more than crystallographic phase and also to include more than one histogram of data in a single refinement. In addition to ferro-, ferri- and commensurate anti-ferromagnetic structures, the new program also allows incommensurate spiral and sine-wave amplitude modulated magnetic structures and their magnetic propagation vectors to be fitted to neutron diffraction data.
An executable program file is available for the Microsoft Windows (W) Intel platform (32 bit, W-95, W-98, W-NT are all tested, w-2000 ?; not alpha or earlier Windows versions). Source code is available upon request and will be offered for inclusion in future versions of CCSL [3].
3. Magnetic structure descriptions.
The CCSL manual describes the input format for Q cards which must be added to the cystal data file, CDF, to define the magnetic structure of a phase. A Q PROP card, which describes the propagation vector of the magnetic structure, must be present if a phase is to be regarded as magnetic by PRODD. The remaining Q cards define the magnetic symmetry and the magnetic moments in magnitude and direction. In incommensurate magnetic structures, the phase angle of a magnetic moment must also be specified. Magnetic form factors are needed along with the rest of the F cards describing a phase. It is recommended that users also prepare input files for the program MAG3D, also part of CCSL, which can display a graphical representation of the magnetic structure described by a complete crystal data file for a single phase. The practice of splitting refinements into separate nuclear and magnetic phases is unnecessary and not recommended.
4. New features of a Crystal Data File (CDF)
Additional features in the CDF not documented in the previous CCSL profile refinement manual include:
|
I ZBAK n |
Indicates whether or not the background should be included in the refinement (n=1) or excluded (n=0). If the is no ZBAK term on the I card then ZBAK 0 is assumed. |
|
F *Sn Label |
Form factors for each source of data have to be listed with n indicating which histogram this form factor is to be applied to in a multi-pattern fit. |
|
L SORC
L SCAL x1 x2 ... |
This card describes the type of diffraction experiment for each histogram (source) of data. Current possibilities are: TF – time of flight neutron diffraction data CN – constant wavelength neutron diffraction data LX – Laboratory x-ray data. Mainly untested, but eventually should account for the Ka1/2 doublet. SR – Synchrotron radiation. Yet to be tested. At the present state of program development, the L SORC information is mainly used to distinguish between angular dispersive and ToF diffraction. As a consequence, one is able to fit a pattern obtained with monochromatic synchrotron radiation by using L SORC CN and x-ray form factors The scale factors x1, x2 to be applied to each dataset are all specified on a single card, in the sequence *S1 *S2 ... etc. |
|
L PKCN *Sn TYPE m |
Indicates which peak-centre function m is to be used for the source n. Currently the only option is 1 for each case. |
|
L PKFN *Sn TYPE m |
Indicates which peak-shape function m is to be used for this phase and source, n. The suite of peak shapes which are currently available are: |
|
TF: 2 Voigt convoluted with double exponential decay |
|
|
TF: 4 As 2 with an extra (L POWR) parameter for the variation of the exponential coefficients with ToF TF: 12 As 2 but allowing MSIG and MGAM to be defined for the magnetic Bragg peaks differently from nuclear peaks (SIGM, GAMM) |
|
|
CN: 1 Gaussian with Cagliotti (U,V,W) width variation [4] CN: 3 Voigt with vL&Y asymmetry correction [5] CN: 4 Pseudo-Voigt with asymmetry correction, based on source code kindly made available by Larry Finger [6] |
|
|
LX: 1 As CN 1. |
|
|
LX: 2 Voigt, no asymmetry. |
|
|
L REFI WORD |
WORD indicates the type of refinement for this phase. The earlier integer system for coding peak shapes and functions will no longer be understood. Possible WORDs are: RIET for conventional Rietveld refinement CAIL for Pawley refinement. (Cell And Intensity Least squares) SAPS for Structure And Peak Shape refinement APES Only RIET and CAIL have been tested so far. The latter can generate a non-positive definite matrix (error) for very heavy peak overlap. |
5. Refinement Control
As in earlier versions of CCSL least-squares refinement, the choice of variables to be refined is made using the instructions L VARY parameter name, L FIX parameter name, L RELAte parameter names and L FUDGe parameter name cards in the CDF of the appropriate phase. However, in the case of parameters which can apply to more than one phase or data histogram, either *Pn or *Sn must precede the parameter name, where n is the number of the phase or histogram.
6. Running PRODD
The component files required by PRODD are a CDF and possibly one or more histogram files (clearly, pattern profile simulation does not require any observed data, with NCYC 0). As with previous versions, the standard data file format is ToF or 2q, intensity and it's esd in free format.
When run, PRODD interactively asks for the name to be used for the output listing file (default extension .LIS) followed by the name of the CDF (default .CCL).
In a magnetic structure refinement, PRODD next asks for a minimum d-spacing for magnetic peaks each time it runs in order to speed up refinements of data which extend to very low d-spacings where magnetic scattering is negligible (eg, the backscattering banks on POLARIS). To include all magnetic peaks enter 0.
Finally, PRODD requires the names of the histogram data files which must be entered in the order * S1, * S2 etc.
The program then proceeds to complete the required number of cycles of refinement unless, at some earlier stage, the refinement converges or the least-squares matrix becomes ill- conditioned. It will also ask for names for any optional files (.HKL, .PRO, .FOU etc) that have been specified and, before beginning the last cycle, for the name of the new crystal data file ( default extension .CCN) to which the new values of the parameters are to be written. A recent development within CCSL is that the estimated standard deviations of positional and unit cell parameters can be written to the .CCN file in the form of an A SD atom name, esd(x), esd(y), esd(z), esd(ITF), esd(site) and similarly for a C SD card.
7. Examining the profile fit
Assuming an I card with an appropriate value for PRPR has been supplied then the program will write out a .PRO file, containing the observed and calculated data. The format of the file is lines containing x back obs calc esd where x is the ToF or 2q specified in the input data file for each obs and esd. For profile fits to more than one histogram of data, the .PRO file contains each fit in the same sequential order as the data files were supplied, with a couple blank lines separating them. Currently it falls to the user to create plots of this data, using a scientific graphing package such as Kaleidagraph. Calculated positions of hkl reflection markers can be included in the .LIS file through the I PRFC card. The .LIS file also contains information from each least-squares cycle giving the new, esd, shift, old and shift/esd values for each variable in the refinement.
For example to plot a Rietveld fit of multi-pattern data using the free "gnuplot" program (Reference????????? ) use the command:
Plot "file.pro" index 0 using 1:3 with dots, "file.pro" index 0 using 1:4 with lines, "file.pro"index 0 using 1:($3-$4) with lines
The x-axis is the ToF or 2q value, the index 0 refers to the first histogram, use index 1, 2 etc to examine the subsequent histograms. Reflection marker positions can be added if the PRFC option was set on the I CARD in the refinement. Taking the list of reflections from the .LIS file gives a set of points which can be over plotted with gnuplot (or any other package).
8. Examples
A number of examples of refinements carried out using PRODD form an Appendix to this document.
9. Installation from source code
Assuming a mistre.ss file and the perl scripts ccsl and get_split have been obtained then executables can be built on a system with working perl and fortran installations. Extraction is a two stage process, the script ccsl takes the mistre.ss file and performs parameter substitutions for array dimensions to suit the user (see jbfpar.mk4) and then get_split separates the large output files into individual fortran subroutines and main programs. To build PRODD one needs to extract the pr, pf, pma and lib sections of the mistre.ss file. The directory structure which is assumed below and in the supplied makefile places the mistre.ss in the root directory, extracted fortran in ./extract and the sections lib, pr etc in directories ./lib, ./pr etc. To use the ccsl script, issue the commands….
perl ccsl lib lax unix ral ./ ./extract/ mistre.ss jbfpar.mk4
perl ccsl pr lax unix ral ./ ./extract/ mistre.ss jbfpar.mk4
perl ccsl pf lax unix ral ./ ./extract/ mistre.ss jbfpar.mk4
perl ccsl pma lax unix ral ./ ./extract/ mistre.ss jbfpar.mk4
which should create the files libmk4.f, prmk4.f, pfmk4.f and pmamk4.f in a directory called extract, using the values in jbfpar.mk4 for parameter substitutions. These large files can either be compiled directly or split up by get_split. Eg
perl get_split split lib ./extract/ ./lib/
This splits the file extract/libmk4.f into individual subroutines in a directory called lib. Each subroutine needs to be compiled and the main program prodd.f is found in the pma section of the mistre.ss files. Rather than splitting the entire file pmamk4.f the source can be extracted with the get mode of the get_split script.
perl get_split get pma ./extract/ ./pma/
The file prodd.got will be found in the pma directory (for many compilers it may be necessary to rename this to prodd.f). Compilation of the Fortran source code is system dependant, we give two examples here.
9.1 Compaq Visual Fortran running under MSWindows.
9.2 G77 running under MSWindows.
A popular free compiler available for a variety of systems, these instructions should be appropriate for the majority of unix systems as well as windows, possibly with minor modifications to the makefile.
Acknowledgements
Alex Hannon for collecting the GEM data (A3), Steve Hull for the CuBr multiphase example (A2)
References
[1] CCSL Website
[2] W I F David, R M Ibberson Ibberson & J C Matthewman (1992) Rutherford Appleton Laboratory Report RAL Report 92-032.
[3] PRODD Website
[4] Cagliotti
[5] Voigt with vL& Y asymmetry correction.
[6] L Finger J. Appl. Cryst. 27, 892, 1992.
[7] Pawley refinement CAILS
[8] Kaleidagraph
[9] gnuplot
APPENDIX
The following examples are designed to illustrate both the differences and the similarities between the input file formats for previous CCSL-based refinement programs and the program PRODD. The structure of the input file is still very similar to the format required for the program MULTI, which was able to perform multi-phase refinement [2]. Cards containing the instructions concerning the number of least-squares cycles to be performed and printing control information are placed at the beginning of the crystal data file. These are followed by cards describing the instrumental parameters for each histogram in a block which starts with a L PKCN *Sn TYPE m card. A further block of cards in the input file then describes each crystallographic phase present in the sample. These blocks are separated by a line beginning with **. These blocks are referred to as phase 0, phase 1, etc – where phase 0 contains the instrumental information.
A1 HRPD Silicon Standard
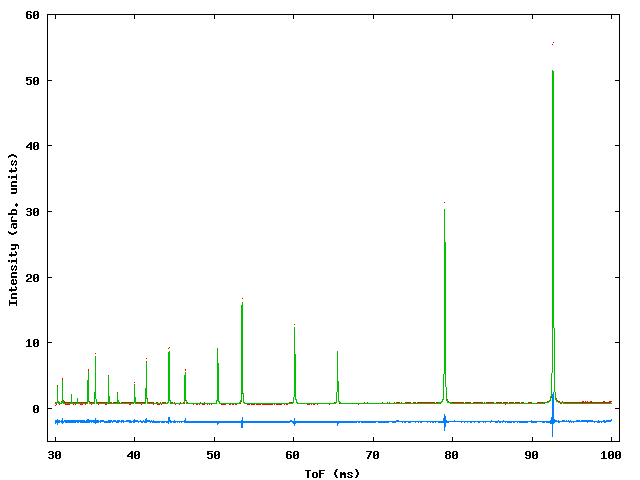
A simple ToF refinement using the silicon standard sample measured on the HRPD instrument at the spallation neutron source ISIS. Two data sets are being refined, histogram 1 (*S1) is from the 90° bank and histogram 2 (*S2) is from the backscattering detector. The parameters describing the shape of the moderator pulse are constrained to be the same for both histograms and are described by the TAUS, TAUF and SWCH parameters. Only a single phase (silicon) is included in this example. Figure 1 shows the fit obtained and table 1 shows the input file along with explanatory comments.

Figure A1: Profile fit to HRPD silicon standard for the backscattering bank (upper) and 90° bank (lower). Red dots – observed data, green line – calculated, blue line – difference.
Table A1 Commented listing of the CDF which produced the profile for Example A1
|
I NCYC 5 MCOR 0 PRIN 0 PRFO 0 PRPR 2 PRFC 2 ZBAK 1 |
Information card. ZBAK is new to PRODD |
|
L SORC TF TF |
Type of data for sources 1,2,3 etc |
|
L SCAL -0.78654 -0.75292 |
Scales for each data source are listed sequentially |
|
Z 90 degree bank |
Comment line |
|
L PKCN *S1 TYPE 1 |
Peak centre type for source 1 |
|
L PKCN 96.7879 0.0000 |
| |
|
L THE2 90.000 |
| |
|
L RTYP 3 20500.00 100000.00 |
| These lines (following PKCN) describe source 1 also |
|
L BACK 2 -2.46074 -6.43948 -4.26476 -2.92025 |
| |
|
L BACK -1.08341 -0.42602 |
| |
|
L ZERO 8.0000 |
| |
|
Z Backscattering.... |
Comment line |
|
L PKCN *S2 TYPE 1 |
Peak centre type for source 2 |
|
L PKCN 95.8864 0.0000 |
| |
|
L RTYP 3 20500.00 100000.00 |
| |
|
L THE2 168.329 |
|These lines describe source 2 |
|
L BACK 2 0.83164 0.02183 |
\ |
|
L ZERO 8.0000 Z |
\ |
|
L VARY ONLY *S1 SCAL |
Fix/vary requests for instrument parameters |
|
L VARY *S2 SCAL |
Constraints concerning more than one phase should be |
|
L VARY *S1 ALL BACK |
placed here. |
|
L VARY *S2 ALL BACK |
|
|
ZL VARY *S1 PKCN 1 |
|
|
ZL VARY *S2 PKCN 1 |
|
|
** |
End of phase 0, start of phase 1 . |
|
N Silicon standard (NBS 640b) |
Title line for phase 1 |
|
Z.. RUN 15610 Cycle 4/97 |
|
|
C 5.430940 5.430940 5.430940 90.0 90.0 90.0 |
Cell parameters |
|
A Si 0.12500 0.12500 0.12500 0.27454 |
Atomic positions (only one required here) |
|
F *S1 Si 1 0.41490 F *S2 Si 1 0.41490 |
Scattering length (note the addition of *Sn) |
|
Z FD-3M no. 227 |
Comment line |
|
S 3/4-X, 1/4-Y, 1/2+Z |
| |
|
S 1/4-X, 1/2+Y, 3/4-Z |
| |
|
S Z, X, Y |
| Symmetry cards giving the generators for Fd-3m |
|
S 3/4+Y, 1/4+X, 1/2-Z |
| |
|
S –X, -Y, -Z |
| |
|
Z |
|
|
L REFI RIET |
Integer values for L REFI cards no longer allowed |
|
L PKFN *S1 TYPE 2 |
Peak function for phase 1 histogram 1 (2 q=90° ) type 2 |
|
L PKFN TAUF 8.0000 0.0000 3.6451 |
| |
|
L PKFN TAUS 8.0000 31.2096 2.0009 |
| |
|
L PKFN SIGM 5.0000 0.0000 196.3015 0.0001 |
| Parameters as described for TF12LS |
|
L PKFN GAMM 20.0000 0.0000 1.1144 0.0001 |
| |
|
L PKFN SWCH 3.0000 4.6516 0.6920 |
| |
|
L PKFN *S2 TYPE 2 |
Peak function for phase 1 source 2 (Backscattering) |
|
L PKFN TAUF 8.0000 0.0000 3.6451 |
|
|
L PKFN TAUS 8.0000 31.2096 2.0009 |
|
|
L PKFN SIGM 5.0000 0.0000 56.5688 0.0001 |
|
|
L PKFN GAMM 20.0000 0.0000 1.2015 0.0001 |
|
|
L PKFN SWCH 3.0000 4.6516 0.6920 |
|
|
Z |
|
|
L VARY *S1 SIGM 2 |
Fix vary requests for peak shape, atomic and unit cell |
|
L VARY *S2 SIGM 2 |
parameters for this phase |
|
ZL VARY *S1 GAMM 2 |
|
|
ZL VARY *S2 GAMM 2 |
|
|
ZL VARY *S1 TAUS 1 *S2 TAUS 1 |
|
|
ZL RELA 1 1.0 *S1 TAUS 1 *S2 TAUS 1 |
Keep the moderator contribution the same for both |
Table 1: Input file for NBS standard silicon on HRPD.
A2 Polaris multiphase fit to high pressure CuBr phases (S. Hull)
A multiphase refinement of POLARIS ToF neutron data for a sample of CuBr in a clamped pressure cell. This fit was originally carried out using the old CCSL based program ‘MULTI’, we reproduce it here to demonstrate the necessary changes to the crystal data file.
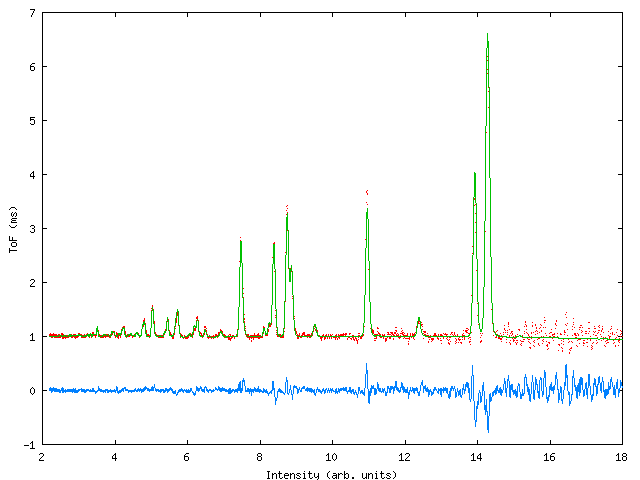
Figure 2: Three phase fit to POLARIS data.
Crystal data file for example A2 (CuBr phases).
Z Multiphase refinement of CuBr #10128a Phase 0
I NCYC 5 MCOR 0 PRIN 0 PRPR 2 PRFC 2 ZBAK 1
L SORC TF
L SCAL 51.450
L PKCN *S1 TYPE 1
L PKCN 12.6013 -1.3654
L RTYP 3 2190.00 18000.00
L WGHT 3
L BACK 2 0.97487 -0.03076 -0.02201 -0.01353
L THE2 90.0
L ZERO -0.7639
Z
L RELA 2 1.0 *P1 SPHA 1.0 *P2 SPHA 1.0 *P3 SPHA
L VARY ONLY SCAL ALL BACK
**
N CuBr Antilitharge
Z
A Cu 0.25000 0.75000 0.00000 2.63032
A Br 0.25000 0.25000 0.29596 1.03770
F Cu 1 0.77180
F Br 1 0.67950
C 3.941062 3.941062 5.005358 90.0 90.0 90.00
Z Space group P4/n (85).....
S 1/2-X, 1/2-Y, Z
S 1/2-Y, X, Z
S -X, -Y, -Z
Z
L REFI RIET
L SPHA 0.66316
L PKFN *S1 TYPE 2
L PKFN TAUF 10.0000 0.0000 0.5564
L PKFN TAUS 10.0000 11.7024 2.4732
L PKFN SWCH 2.0000 0.3194 0.0000
Z
Z Sample parameters.....
L PKFN SIGM 5.0000 0.0000 192.9444 -5.6319
L PKFN GAMM 20.0000 0.0000 2.7509 0.0000
Z
ZL RELA 1 1.0 Cu ITF 1.0 Br ITF
Z
Z Variable parameters.....
L VARY ONLY SPHA
L VARY ALL CELL
L VARY ALL ITF
L VARY SIGM 2 SIGM 3
L VARY GAMM 2
L VARY ALL XYZ
**
N CuBr Zincblende
Z
A Cu 0.00000 0.00000 0.00000 4.44242
A Br 0.25000 0.25000 0.25000 4.44242
F Cu 1 0.77180
F Br 1 0.67950
C 5.498016 5.498016 5.498016 90.00000 90.00000 90.00000
Z Space Group = F-43m.....
S X, Y+1/2, Z+1/2
S -X, -Y, Z
S -X, Y, -Z
S Z, X, Y
S Y, X, Z
Z
Z Refinement details.....
L REFI RIET
L SPHA 0.32731
L PKFN *S1 TYPE 2
L PKFN TAUF 10.0000 0.0000 0.5564
L PKFN TAUS 10.0000 11.7024 2.4732
L PKFN SWCH 2.0000 0.3194 0.0000
Z
Z Sample parameters.....
L PKFN SIGM 5.0000 0.0000 188.9911 -3.6212
L PKFN GAMM 20.0000 0.0000 2.8303 0.0000
Z
L RELA 1 1.0 Cu ITF 1.0 Br ITF
Z
Z Variable parameters.....
L VARY ONLY SPHA
L VARY ALL CELL
L VARY ALL ITF
L VARY SIGM 2 SIGM 3
L VARY GAMM 2
**
N CuBr new phase
Z
A Br 0.15338 0.15338 0.15338 3.58357
A Cu 0.62771 0.62771 0.62771 3.52462
F Cu 1 0.77180
F Br 1 0.67950
C 6.742560 6.742560 6.742560 90.0 90.0 90.00
Z Space group Pa3
S -X+1/2, -Y, Z+1/2
S -X, Y+1/2, -Z+1/2
S Z, X, Y
S -X, -Y, -Z
Z
Z Refinement details.....
L REFI RIET
L SPHA 0.00955
L PKFN *S1 TYPE 2
L PKFN TAUF 10.0000 0.0000 0.5564
L PKFN TAUS 10.0000 11.7024 2.4732
L PKFN SWCH 2.0000 0.3194 0.0000
Z
Z Sample parameters.....
L PKFN SIGM 5.0000 0.0000 93.5900 0.0000
L PKFN GAMM 20.0000 0.0000 2.0000 0.0000
Z
L RELA 1 1.0 Cu ITF 1.0 Br ITF
Z
Z Variable parameters.....
L VARY ONLY SPHA
L VARY ALL CELL
L VARY ALL ITF
L VARY SIGM 2
A3 Weighed multiphase, multi-histogram magnetic sample on GEM
Data were collected for sample containing a mixture of NiO, CeO2 and NaCl in weighed proportions to test the capabilities of the program with respect to determination of phase fractions in the mixture. At the time of the experiment the commissioning of the GEM instrument was in progress and three data banks were available, giving ToF data from detectors centered around 2q values of 90° , 70° and 20° . Figure 3 shows the profile fit to the data., table 3 gives the input file.
The refined values of SPHA for NiO, CeO2 and NaCl were 0.3312(4), 0.3329(5) and 0.3360(7) respectively. These reflect the relative number of each kind of unit cell in the sample. By looking at the site multiplicities in the .lis file, the number of each kind of atom in the unit cell can be calculated and hence the mass of the unit cell. The weight and atomic fractions are then simply:
wt. fraction =  at. fraction =
at. fraction = 
where Mcell is the mass of the unit cell. For this example the SPHA values have been constrained such that they sum to 1.0 and there are equal numbers of formula units in the unit cells of each phase. Hence, the atomic fractions are just the SPHA values and the wt fractions are calculated as: NiO = 24.33(3)% , CeO2 = 56.34(6) and NaCl = 19.33(4). These favourably with the measured values of 24.6%, 56.2% and 19.1%, with due uncertainty in the purity of the starting materials and the presence or otherwise of amorphous phases.
The input file for this fit is shown below. It is rather long, but the main new feature is the addition of a group of Q cards to describe the magnetic structure in NiO. See the CCSL manual [] for further details relating to Q cards. The refined moment of 1.840(8) mB is in good agreement with the value expected for Ni2+, allowing for covalency and zero point spin deviation.

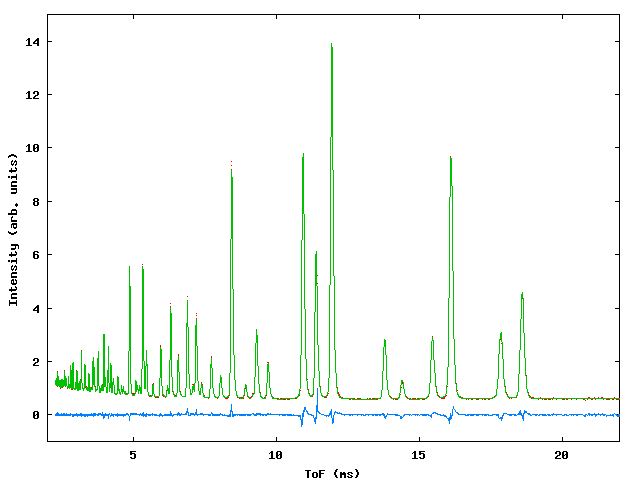
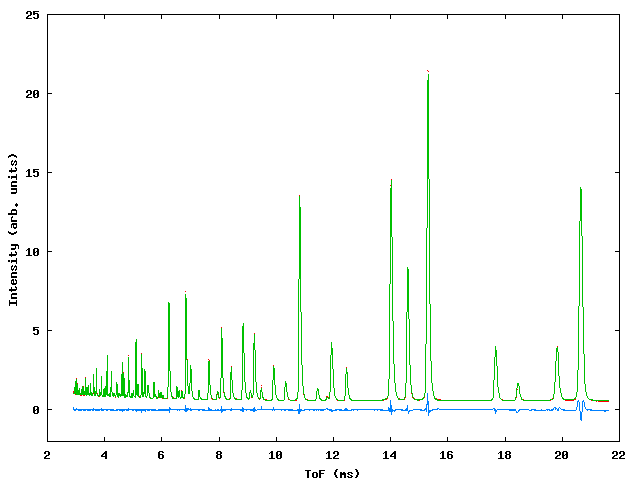
Figure 3: Profile fits to the GEM multiphase sample. Top left: 20° bank, top right 70° bank, bottom: 90° bank.
Z Multiphase sample GEM NiO/CeO2/NaCl
Z
I NCYC 5 MCOR 0 PRIN 0 PRFO 0 PRPR 2
I PRRC 0 ZBAK 1
L SORC TF TF TF
L SCAL 0.1390E+07 0.122E+07 0.7005E+06
Z
Z GEM 90 deg
L PKCN *S1 TYPE 1
L PKCN 18.3556 -0.2108
L THE2 103.876
L RTYP 3 2920.00 21500.00
L ABSC 1 0.0000 0.0108
L BACK 5 2500.00000 1.41482
L BACK 3000.00000 0.95037
L BACK 3500.00000 0.88858
L BACK 4000.00000 0.83244
L BACK 5000.00000 0.74138
L BACK 7000.00000 0.64603
L BACK 10000.00000 0.58047
L BACK 15000.00000 0.57543
L BACK 25000.00000 0.59865
L ZERO -0.3004
Z
Z GEM 70 deg
L PKCN *S2 TYPE 1
L PKCN 18.0014 1.3416
L THE2 77.4389
L ABSC 1 0.0000 -0.0094
L RTYP 3 2274.00 22000.00
L BACK 5 2200.00000 1.04500
L BACK 2900.00000 0.92383
L BACK 3500.00000 0.84077
L BACK 4000.00000 0.79134
L BACK 5000.00000 0.70090
L BACK 7000.00000 0.62623
L BACK 10000.00000 0.58867
L BACK 15000.00000 0.58212
L BACK 25000.00000 0.62512
L ZERO 2.9087
ZZ
Z GEM 20 deg
L PKCN *S3 TYPE 1
L PKCN 18.5026 -3.7339
L THE2 21.7693
L ABSC 1 0.0000 -0.4561
L RTYP 3 1800.00 10000.00
L BACK 5 1500.00000 0.94131
L BACK 2000.00000 0.85832
L BACK 3000.00000 0.70724
L BACK 5000.00000 0.57864
L BACK 7500.00000 0.56957
L ZERO -1.0819
Z
L VARY *S1 SCAL 1
L VARY *S2 SCAL 1
L VARY *S3 SCAL 1
L VARY *S1 ALL BACK *S2 ALL BACK *S3 ALL BACK
L VARY *S1 ABSC 2 *S2 ABSC 2 *S3 ABSC 2
L VARY *S2 PKCN 1 *S2 PKCN 2
L RELA 2 1.0 *P1 SPHA 1.0 *P2 SPHA 1.0 *P3 SPHA
L RELA 1 1.0 *P1 *S1 TAUS 1 1.0 *P2 *S1 TAUS 1
L RELA 1 1.0 *P1 *S1 TAUS 1 1.0 *P3 *S1 TAUS 1
L RELA 1 1.0 *P1 *S1 TAUS 2 1.0 *P2 *S1 TAUS 2
L RELA 1 1.0 *P1 *S1 TAUS 2 1.0 *P3 *S1 TAUS 2
**
N NiO from NiCO3.xH2O (1000 degC) GEM00735
A Ni 0.000 0.000 0.000 0.33566
A O 0.500 0.500 0.500 0.40713
F *S1 Ni 1 1.0300
F *S1 O 1 0.5803
F *S1 NiM 2 -.0172 35.739 .3174 14.269 .7136 4.566 -.0143
F *S2 Ni 1 1.0300
F *S2 O 1 0.5803
F *S2 NiM 2 -.0172 35.739 .3174 14.269 .7136 4.566 -.0143
F *S3 Ni 1 1.0300
F *S3 O 1 0.5803
F *S3 NiM 2 -.0172 35.739 .3174 14.269 .7136 4.566 -.0143
C 4.183181 4.183181 4.183181 90.0 90.0 90.0
Z Fm-3m no. 225
S GRUP F M 3 M
Q STYP ANTI
Q PROP 0.5000 0.5000 0.5000
Q NiM FORM Ni
Q MU 1.8412
Q SDIR 35.2644 45.0000
Q MSYM -1 1 5 1
Q NSYM 2 1 0 0 0 1 0 0 0 1
Q NSYM 3 1 0 0 0 1 0 0 0 1
Q NSYM 4 1 0 0 0 1 0 0 0 1
Q NSYM 6 1 0 0 0 1 0 0 0 1
Q NSYM 8 1 0 0 0 1 0 0 0 1
Q NSYM 14 1 0 0 0 1 0 0 0 1
Q NSYM 15 1 0 0 0 1 0 0 0 1
Z
L REFI RIET
L PKFN *S1 TYPE 2
L PKFN TAUF 8.0000 0.0000 4.4704
L PKFN TAUS 8.0000 32.7580 1.7429
L PKFN SIGM 15.0000 0.0000 41.9703 1.1362
L PKFN GAMM 20.0000 0.0000 3.6262 -0.0644
L PKFN SWCH 3.0000 4.1919 0.6578
Z
L PKFN *S2 TYPE 2
L PKFN TAUF 8.0000 0.0000 4.4704
L PKFN TAUS 8.0000 32.7580 1.7429
L PKFN SIGM 15.0000 0.0000 124.5899 1.8183
L PKFN GAMM 20.0000 0.0000 5.4009 0.1140
L PKFN SWCH 3.0000 4.1919 0.6578
Z
L PKFN *S3 TYPE 2
L PKFN TAUF 8.0000 0.0000 4.4704
L PKFN TAUS 8.0000 32.7580 1.7429
L PKFN SIGM 15.0000 0.0000 3625.4126 0.0001
L PKFN GAMM 20.0000 0.0000 17.6227 0.0001
L PKFN SWCH 3.0000 4.1919 0.6578
Z
L VARY ALL ITF
L SPHA 0.32753
L VARY SPHA
L VARY Ni MU A*
L VARY *S1 SIGM 2 *S1 GAMM 2 *S1 GAMM 3
L VARY *S2 SIGM 2 *S2 GAMM 2 *S2 GAMM 3
L VARY *S3 SIGM 2 *S3 GAMM 2
L VARY *S1 TAUS 1 *S2 TAUS 1 *S3 TAUS 1
L RELA 1 1.0 *S1 TAUS 1 1.0 *S2 TAUS 1
L RELA 1 1.0 *S1 TAUS 1 1.0 *S3 TAUS 1
L VARY *S1 TAUS 2 *S2 TAUS 2 *S3 TAUS 2
L RELA 1 1.0 *S1 TAUS 2 1.0 *S2 TAUS 2
L RELA 1 1.0 *S1 TAUS 2 1.0 *S3 TAUS 2
**
N CeO2 from Chemical Cupboard (??) GEM00735
A Ce 0.00000 0.00000 0.00000 0.35024 0.01
A O 0.25000 0.25000 0.25000 0.51020 0.01
F *S1 Ce 1 0.4840
F *S1 O 1 0.5803
F *S2 Ce 1 0.4840
F *S2 O 1 0.5803
F *S3 Ce 1 0.4840
F *S3 O 1 0.5803
C 5.418159 5.418159 5.418159 90.0 90.0 90.0
Z Fm-3m no. 225
S GRUP F M 3 M
Z
L REFI RIET
L PKFN *S1 TYPE 2
L PKFN TAUF 8.0000 0.0000 4.4704
L PKFN TAUS 8.0000 32.7580 1.7429
L PKFN SIGM 15.0000 0.0000 36.1599 1.1362
L PKFN GAMM 20.0000 0.0000 4.5488 0.4939
L PKFN SWCH 3.0000 4.1919 0.6578
Z
L PKFN *S2 TYPE 2
L PKFN TAUF 8.0000 0.0000 4.4704
L PKFN TAUS 8.0000 32.7580 1.7429
L PKFN SIGM 15.0000 0.0000 119.2410 1.8183
L PKFN GAMM 20.0000 0.0000 5.7774 0.8615
L PKFN SWCH 3.0000 4.1919 0.6578
ZZ
L PKFN *S3 TYPE 2
L PKFN TAUF 8.0000 0.0000 4.4704
L PKFN TAUS 8.0000 32.7580 1.7429
L PKFN SIGM 15.0000 0.0000 2460.1448 0.0001
L PKFN GAMM 20.0000 0.0000 41.7493 0.0001
L PKFN SWCH 3.0000 4.1919 0.6578
Z
L SPHA 0.32902
L VARY SPHA
L VARY A* ALL ITF
L VARY *S1 SIGM 2 *S1 GAMM 2 *S1 GAMM 3
L VARY *S2 SIGM 2 *S2 GAMM 2 *S2 GAMM 3
L VARY *S3 SIGM 2 *S3 GAMM 2
L VARY *S1 TAUS 1 *S2 TAUS 1 *S3 TAUS 1
L RELA 1 1.0 *S1 TAUS 1 1.0 *S2 TAUS 1
L RELA 1 1.0 *S1 TAUS 1 1.0 *S3 TAUS 1
L VARY *S1 TAUS 2 *S2 TAUS 2 *S3 TAUS 2
L RELA 1 1.0 *S1 TAUS 2 1.0 *S2 TAUS 2
L RELA 1 1.0 *S1 TAUS 2 1.0 *S3 TAUS 2
**
N NaCl from Chemical Cupboard (??) GEM00735
A Na 0.00000 0.00000 0.00000 1.76500 0.01
A Cl 0.50000 0.50000 0.50000 1.35850 0.01
F *S1 Na 1 0.363
F *S1 Cl 1 0.9577
F *S2 Na 1 0.363
F *S2 Cl 1 0.9577
F *S3 Na 1 0.363
F *S3 Cl 1 0.9577
C 5.645169 5.645169 5.645169 90.0 90.0 90.0
Z Fm-3m no. 225
S GRUP F M 3 M
Z
L REFI RIET
L PKFN *S1 TYPE 2
L PKFN TAUF 8.0000 0.0000 4.4704
L PKFN TAUS 8.0000 32.7580 1.7429
L PKFN SIGM 15.0000 0.0000 42.3861 1.1362
L PKFN GAMM 20.0000 0.0000 4.4124 -0.1611
L PKFN SWCH 3.0000 4.1919 0.6578
Z
L PKFN *S2 TYPE 2
L PKFN TAUF 8.0000 0.0000 4.4704
L PKFN TAUS 8.0000 32.7580 1.7429
L PKFN SIGM 15.0000 0.0000 128.4710 1.8183
L PKFN GAMM 20.0000 0.0000 5.2031 0.0676
L PKFN SWCH 3.0000 4.1919 0.6578
ZZ
L PKFN *S3 TYPE 2
L PKFN TAUF 8.0000 0.0000 4.4704
L PKFN TAUS 8.0000 32.7580 1.7429
L PKFN SIGM 15.0000 0.00 3427.7664 0.0001
L PKFN GAMM 20.0000 0.00 25.5840 0.0001
L PKFN SWCH 3.0000 4.1919 0.6578
Z
L SPHA 0.34345
L VARY SPHA
L VARY A*
L VARY ALL ITF
L VARY *S1 SIGM 2 *S1 GAMM 2 *S1 GAMM 3
L VARY *S2 SIGM 2 *S2 GAMM 2 *S2 GAMM 3
L VARY *S3 SIGM 2 *S3 GAMM 2
L VARY *S1 TAUS 1 *S2 TAUS 1 *S3 TAUS 1
L RELA 1 1.0 *S1 TAUS 1 1.0 *S2 TAUS 1
L RELA 1 1.0 *S1 TAUS 1 1.0 *S3 TAUS 1
L VARY *S1 TAUS 1 *S2 TAUS 2 *S3 TAUS 2
L RELA 1 1.0 *S1 TAUS 2 1.0 *S2 TAUS 2
L RELA 1 1.0 *S1 TAUS 2 1.0 *S3 TAUS 2
A4 Rb2S2O6 synchrotron x-ray diffraction at ambient temperature on beamline BM16 at ESRF.
Sharp diffraction profiles are well fitted by peakshape number 4, which is based on the source code of L. Finger[6]. Note that L SORC CN has been used although this is clearly x-ray diffraction data. The Lorentz-Polarisation correction is the same in both instances.


Crystal data file for Rb2S2O6, note the temperature factors are incorrect since no the absorbtion correction has yet to be applied.
I NCYC 6 MCOR 0 FRIE 1 PRIN 0 PRFO 0 PRPR 2 PRFC 0 ZBAK 1
L SCAL 0.20290E-01
L SORC CN
L PKCN *S1 TYPE 1
L RTYP 3 3.000 80.000 0.003
L WGHT 3
L WVLN 0.850418
L BACK 5 3.00000 12.45016
L BACK 25.00000 28.74267
L BACK 50.00000 30.28023
L BACK 80.00000 20.69130
L ZERO -0.0010
Z
L VARY ONLY SCAL 1
L VARY ALL BACK
L VARY ZERO 1
**
N Rb2s2o6 room temperature 297 K structure in P321 from BM16 (ESRF)
C 10.182619 10.182619 6.434160 90.00000 90.00000 120.00000
S -y, x-y, z
S y, x, -z
A Rb1 0.37857 0.00000 0.00000 2.71782
A Rb2 0.70472 0.00000 0.50000 2.71782
A S1 0.00000 0.00000 0.16860 1.18974
A S2 0.33333 0.66667 0.41344 1.18974
A S3 0.33333 0.66667 0.74044 1.18974
A O1 0.85775 0.88422 0.22607 1.46932
A O2 0.79273 0.48081 0.19404 1.46932
A O3 0.80574 0.34609 0.64484 1.46932
F Rb 2 17.1784 1.7888 9.6435 17.3151 5.1399 0.2748 1.5292 164.934 3.48730
F S 2 6.9053 1.4679 5.2034 22.2151 1.4379 0.2536 1.5863 56.172 0.8669
F O 2 3.0485 13.2771 2.2868 5.7011 1.5463 0.3239 0.867 32.9089 0.2508
L REFI RIET
L PKFN *S1 TYPE 4
L PKFN SIGM 10.0000 0.0000 0.0000 0.0001
L PKFN GAMM 25.0000 0.0089 0.0392
L PKFN SOVL 0.0000 0.0003
L PKFN DOVL 0.0000 0.0002
Z
L VARY ALL CELL
L VARY GAMM X GAMM Y
L VARY ALL XYZT
L RELA 1 1.0 Rb1 ITF 1.0 Rb2 ITF
L RELA 1 1.0 S1 ITF 1.0 S2 ITF
L RELA 1 1.0 S1 ITF 1.0 S3 ITF
L RELA 1 1.0 O1 ITF 1.0 O2 ITF
L RELA 1 1.0 O1 ITF 1.0 O3 ITF