
![]()
![]()
Department of Chemistry
University of Nottingham, UK
(Wednesday 10th January 2001)
|
|
Department of Chemistry University of Nottingham, UK (Wednesday 10th January 2001) |
|
"Coping with the Niagara Falls of data and difficult crystallographic problems":
Making the best out of freely available crystallographic software for single crystal and powder diffraction..
Lachlan M.D. Cranswick
Collaborative Computational Project No 14 (CCP14) for Single Crystal and Powder Diffraction
Daresbury Laboratory, Warrington, WA4 4AD U.K
E-mail: L.Cranswick@dl.ac.uk
WWW:
http://www.ccp14.ac.uk
|
|
Freely Available Software tools to aid in Structure Solution from Powder Diffraction Notes on the Web Lachlan M.D. Cranswick (L.Cranswick@dl.ac.uk) CCP14 Project for Single Crystal and Powder Diffraction http://www.ccp14.ac.uk ccp14@ccp14.ac.uk |
|

Hand Written Note Free Zone!!!!
Put down those pens!!
Some areas of this talk may resemble a rather fast computer slide show; thus detailed notes are on the web for examination at your leisure:
http://www.ccp14.ac.uk/poster-talks/nottingham2001/
Link Via:
http://www.ccp14.ac.uk
Single Crystal Methods
(Mass tourism structure solution)

Some Possible Problems:
Crystal not representative of the bulk material
Twinning
Crystal explodes during take-off/data collection
Structure Validation
Cambridge Database Stats:
( http://www.ccdc.cam.ac.uk/about/annrep99/Report.html )
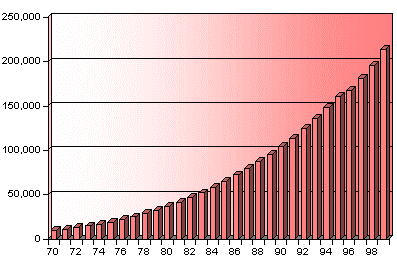
"During 1999, 17,898 new entries were added"
(that Scale is in the 100’s of thousands)
MicroCrystal Beamline 9.8 at Daresbury Lab.
(managed by Simon Teat: http://srs.dl.ac.uk/xrd/9.8/)
(is your powder really just a collection of small crystals?)
82 publications in 3.5 years (35 publications in 1999)
Estimated 1,000 to 1,500
structures solved in that 3.5 years on 9.8 (30 minutes to 12 hours per data collection).Powder Methods
(a nightmare to some? – an Adventure to Others!)
Powder Solving Stats:
( http://sdpd.univ-lemans.fr/iniref.html )
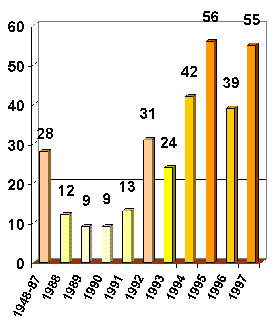
Powder: "During 1999: 55 publications"
( http://sdpd.univ-lemans.fr/iniref/a1999.html )
(a cumulative scale would be in the 100’s)
(Cambridge: "During 1999: 17,898 new entries")
Structure Determination from Powder Diffractometry Round Robin
Tetracycline Hydrochloride:
(June 1998)
http://sdpd.univ-lemans.fr/SDPDRR/
Armel Le Bail and Lachlan Cranswick
Powder Data:
6 week time limit
70 downloads of data
2 submissions on the Tetracycline within the time limit
CSD System from Stoe
http://www.stoe.com/products/
http://imr.chem.binghamton.edu/zavalij/CSD.html
Druid/Mystic (now called Dash)
http://www.esrf.fr/info/science/highlights/1999/chemistry/powder.html
(solved by Armel Le Bail)
http://sdpd.univ-lemans.fr/SDPDRR/sample2.html
Structure Determination from Powder Diffractometry Round Robin
Single Crystal Data?
(a powder can be a collection of small crystals)
10x20x30 micron crystal (Clegg and Teat).
http://srs.dl.ac.uk/xrd/9.8/
"Routine" structure solution within the hour (before all the data was collected)
Bruker Smart CCD: BL 9.8, Daresbury:
Agenda
Which phases are present and has the Structure been Solved Already?
Phase Identification / Search-Match for Powder Diffraction
Crystal Structure Databases
Data Conversion/Importing data/structures
Peak Profiling
Powder Indexing
Unit-Cell Refinement
Powder Diffraction Utility Software
Full Profile Fitting
Materials Analysis Rietveld/Texture Software
Structure Solution (Single Crystal/Powder Diffraction)
Structure Refinement (Single Crystal/Powder Diffraction)
Single Crystal Suites
Fourier Maps
Structure Validation
Specialised Programs (Charge Density/Anharmonic/Incommensurate)
Quantitative Phase Analysis
Powder Diffraction Pattern Calculation
Structure Visualisation and Photorealistic Hardcopy Output
Dual boot Windows and UNIX (Linux or FreeBSD) PCs
Free Xtal Nexus CD-ROMs for students and academics isolated from the internet
Has the structure been found already?
Phase Identification/Search Match for Powder Diffraction
Two main parts to perform computer based search-match:
1. Have a Powder Diffraction Database (buy or make your own)
2. Search-match software that uses the above database to search.
Databases:
ICDD has the commercial powder diffraction database area cornered http://www.icdd.com
Alternative being developed is the Pauling File
Nearly all Search-match programs are commercial:
Refer to, "Available Search-Match Software" for a list of known software:
http://www.ccp14.ac.uk/solution/search-match.htm
Known exceptions being:
Brian Toby's "Portable Logic Program" (UNIX)
http://www.ncnr.nist.gov/programs/crystallography/
"MacDiff" for Apple Mac freeware by Rainer Petschick
http://www.geol.uni-erlangen.de/macsoftware/macdiff/
Phase Identification/Search Match for Powder Diffraction
Identifying an organic – DL-Valine

Multiphase mixture: Flourite, Corundum, Zincite

Has the structure been solved already?
Crystallographic Structure Databases
(UK based academics and students already have free access via the EPSRC funded CDS (Chemical Database Service):
http://cds.dl.ac.uk/
ICSD (Inorganics)
http://www.fiz-karlsruhe.de/
Web accessible demonstration:
http://barns.ill.fr/dif/icsd/
MDF/CRYSTMET
(Metals and Alloys)
http://www.tothcanada.com
CCSD
(Organics and Organometallics)
http://www.ccdc.cam.ac.uk/
ICSD via the Web
Using an Interface created by Alan Hewat
There is a trend for many types of databases to use the web due to the convenience and effectiveness. Also has advantage of being operating system independent.
http://barns.ill.fr/dif/icsd/
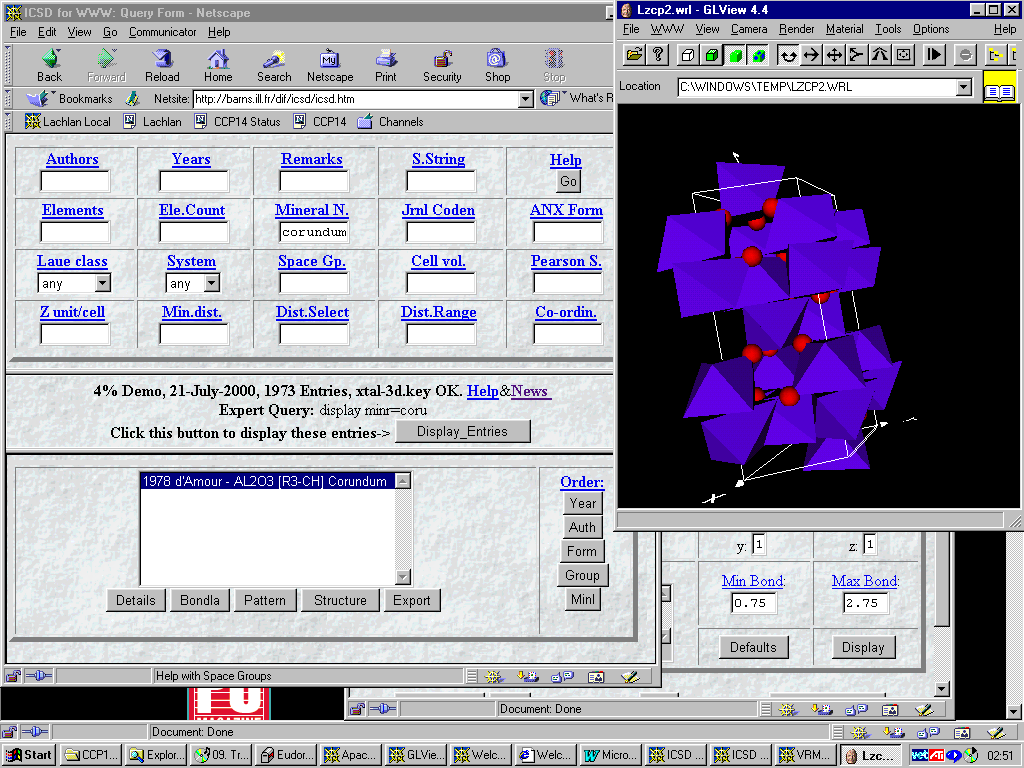
A hopeful new trend -
Crystallography Suites that link directly into structure databases:(Platon/System S and Crystals linking into the Cambridge Database)
Platon/System S as a user-friendly interface into CSD Quest
http://www2.ccc.uni-erlangen.de/software/corina/free_struct.html
Platon:
http://www.crystal.chem.uu.nl/software.html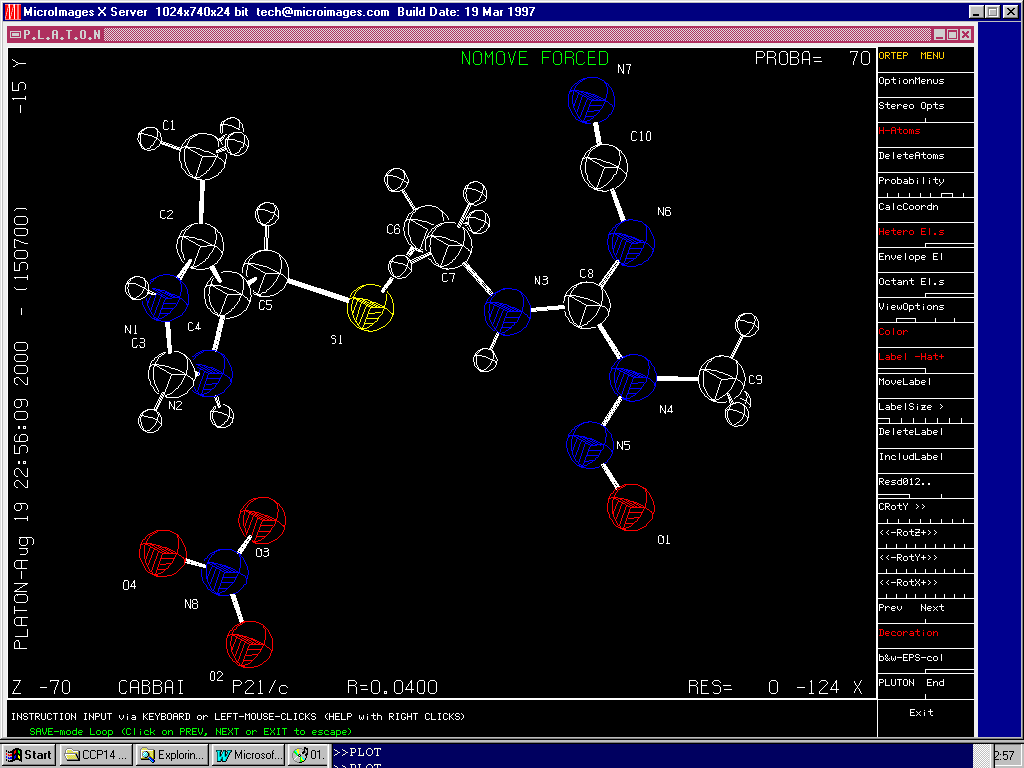
CSD Cell searching
via a point and click interface in Platon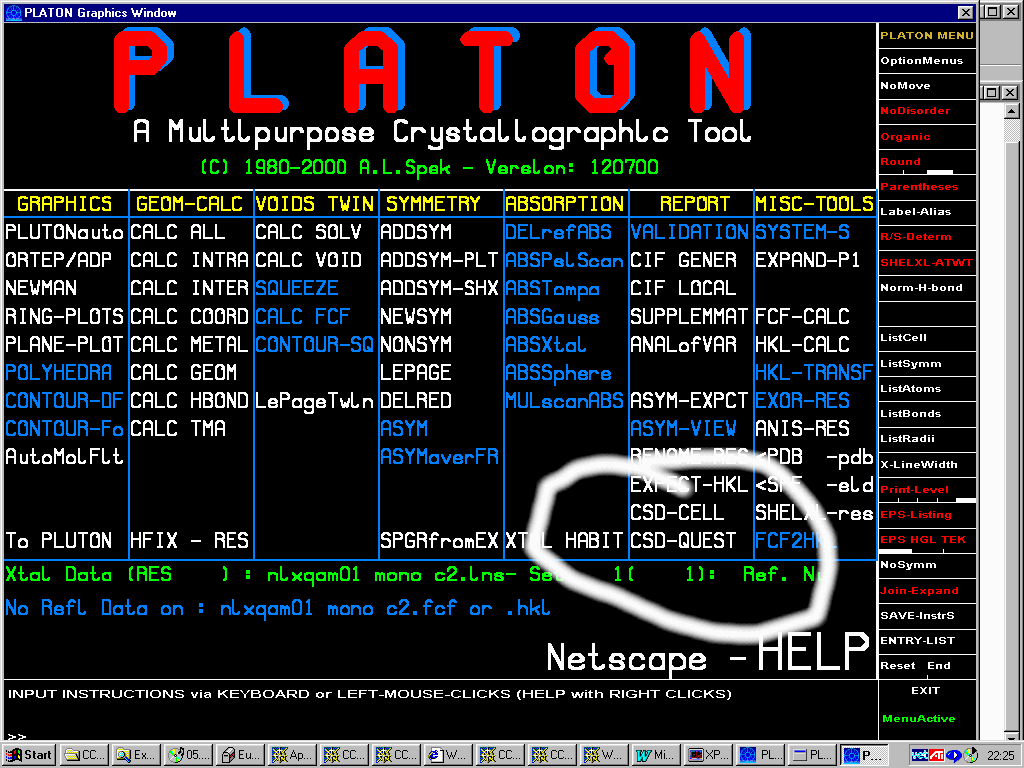
Crystals single Crystal Suite
performing bond-length and bond-angle validation using the Cambridge Database as part of the structure refinement process(still under development)
Crystals: http://www.xtl.ox.ac.uk/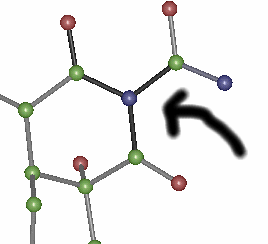
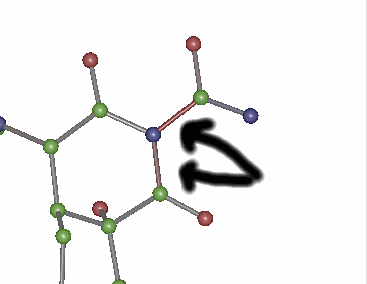
After running auto generated
scripts through quest:
Red bonds imply bonds are
too short for this geometry
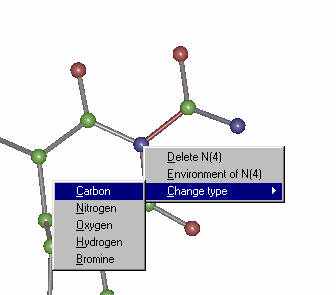
Change Nitrogen to Carbon using the mouse
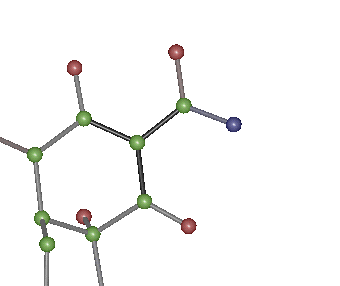
Bonds now within what is
expected from the CSD
Crystals:
Though of course you can look at the thermal ellipsoids (i.e., via Cameron provided with Crystals):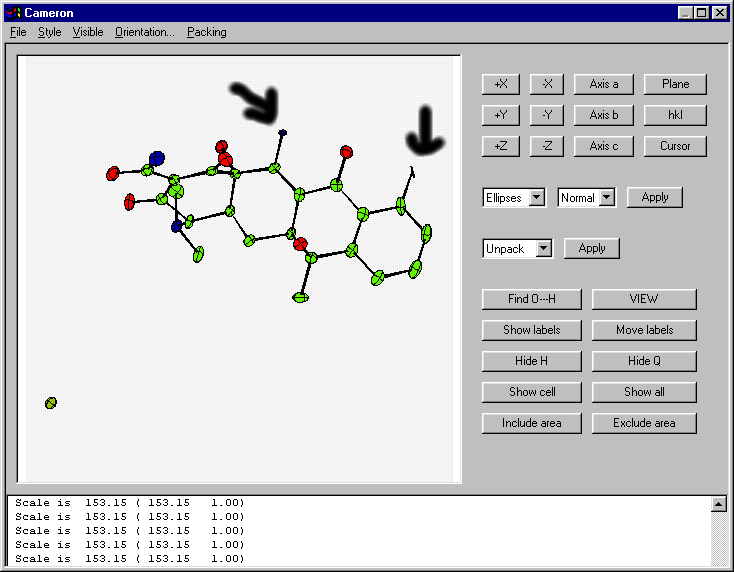
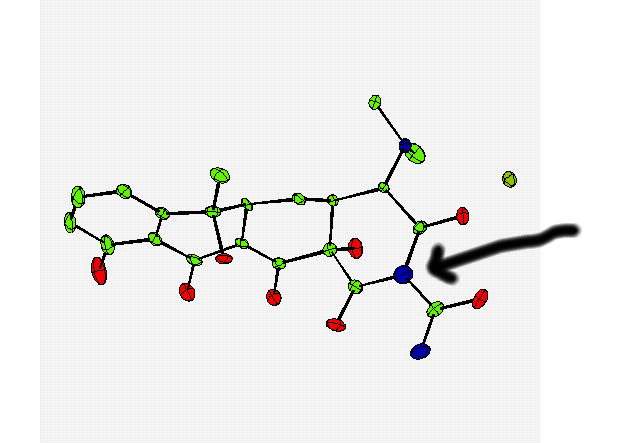
Though in the above validation case (to a novice crystallographer) the thermal from the incorrect atom may not be so obvious.
Powder Data Conversion/Importing Data
Initial problem in powder diffraction can be getting the data in the right format. For interconverting powder diffraction data: a vareity of programs exist which in combination can pretty much get you from one format to another.
Use of a text editor and spreadsheet may be occassionally required.
A very good text editor is the freeware PFE Editor for Windows:
http://www.lancs.ac.uk/people/cpaap/pfe/
Context for Windows (freeware) can do column mode editing:
(toggle Control L for normal and column editing mode)
http://www.fixedsys.com/context/
Refer: Data Conversion/Importing data/structures
http://www.ccp14.ac.uk/solution/powderdataconv/
These Include: ConvX, Powder v2.00, Powder X, Winfit
Example of the ConvX for Windows : mass conversion of powder diffraction data
(http://www.ceramics.irl.cri.nz/Convert.htm):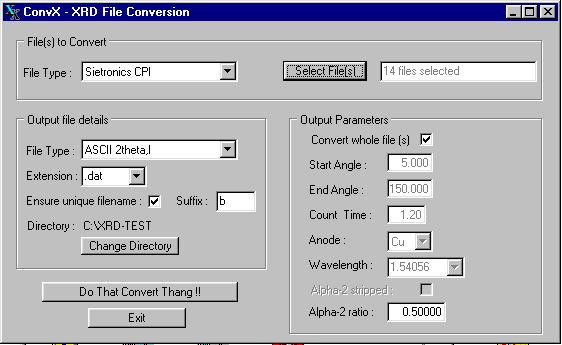
Structure Importing, Conversion and Transformation
Summary List at:
http://www.ccp14.ac.uk/solution/structconv/
Be careful with these and check the results.
Best program at the moment is the shareware program CRYSCON
http://www.shapesoftware.com
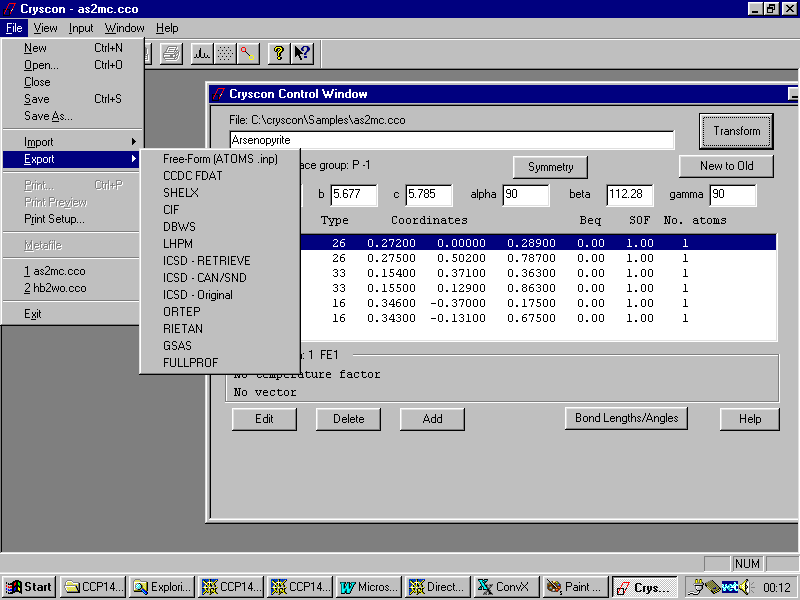
Other software such as Ortep-3 can import a wide variety of formats and output these as Shelx files.
Powder Diffraction Utility Software
Examining Data, peak finding, background stripping, alpha-2 stripping
Powder v 2.00:
http://www.ccp14.ac.uk/tutorial/powder/Powder X,
http://www.ccp14.ac.uk/tutorial/powderx/WinFIT,
http://www.geol.uni-erlangen.de/html/software/soft.htmlWinplotr,
http://www-llb.cea.fr/winplotr/winplotr.htmXFIT,
http://www.ccp14.ac.uk/tutorial/xfit-95/xfit.htmExample of PowderX for Windows
Graphical evaluation, backtground stripping, smoothing, alpha stripping, peak find and pass to treor indexing
Full GUI Operation
Alpha2 Strip, Background Strip, Peak Find
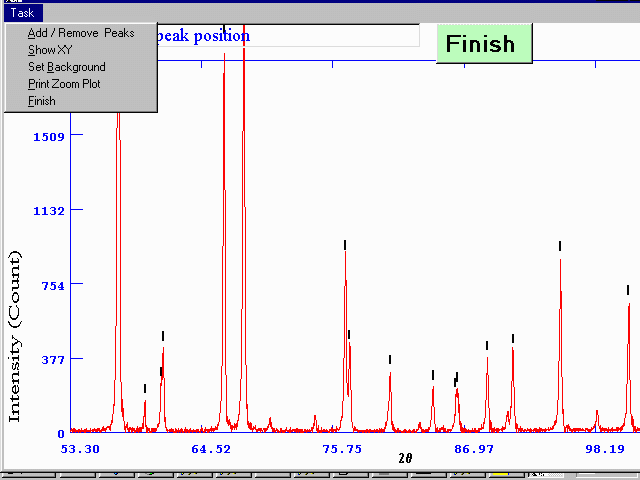
Peak Finding/Peak Profiling
For Overall Summary of available peak profiling software refer to:
http://www.ccp14.ac.uk/solution/peakprofiling/
These include: CMPR, DRXWin, EFLECH, GPLSFT, pearson.xls, SHADOW, Powder v2.00, PowderX, Winfit, Winplotr, XFIT
Example of XFIT for Windows (this can also perform fundamental parameters peak profiling for high accuracy peak positions from laboratory diffractometer data). This can be the difference between a convincing and unconvincing powder indexing from laboratory X-ray data.
Powder Indexing:
A Non-trivial endeavourFor Overall Summary of available powder indexing software refer to:
http://www.ccp14.ac.uk/solution/indexing/
Powder Indexing:
Autox, Ito, Dicvol, Treor, Taup/Powder, Lzon, Losh, Kohl, Scanix, Xrayscan, EFLECH/Index, SupercellLinking Suites:
Crysfire, Powder v2.00, PowderX, PROSZKI, WinPlotr, ChekcellE.g., supercel is a specialise indexing program by Juan Rodriguez-Carvajal for indexing Super Cell and Incommensurate cells. (available within Winplotr)
http://www-llb.cea.fr/winplotr/winplotr.htm
At present the CRYSFIRE software by Robin Shirley links 8 different indexing programs (ito, dicvol, treor, taup, kohl, lzon, fjzn and losh) together with a common interface and using intelligent defaults. Important to have access to as many indexing programs as possible so you can get a feel for the range of possible solutions.
http://www.ccp14.ac.uk/tutorial/crys/
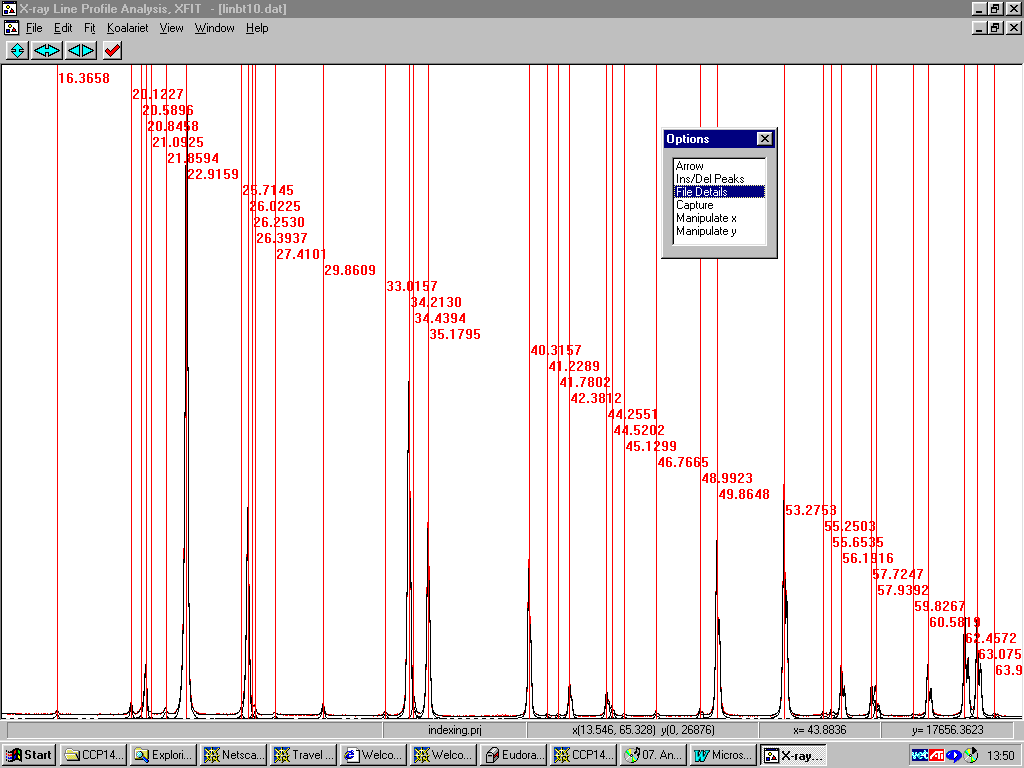
Example of CRYSFIRE Screen prompting the saving into one of 8 different indexing program formats:
Interpreting Crysfire Summary Files:
Powder Indexing and Spacegroup Assignment
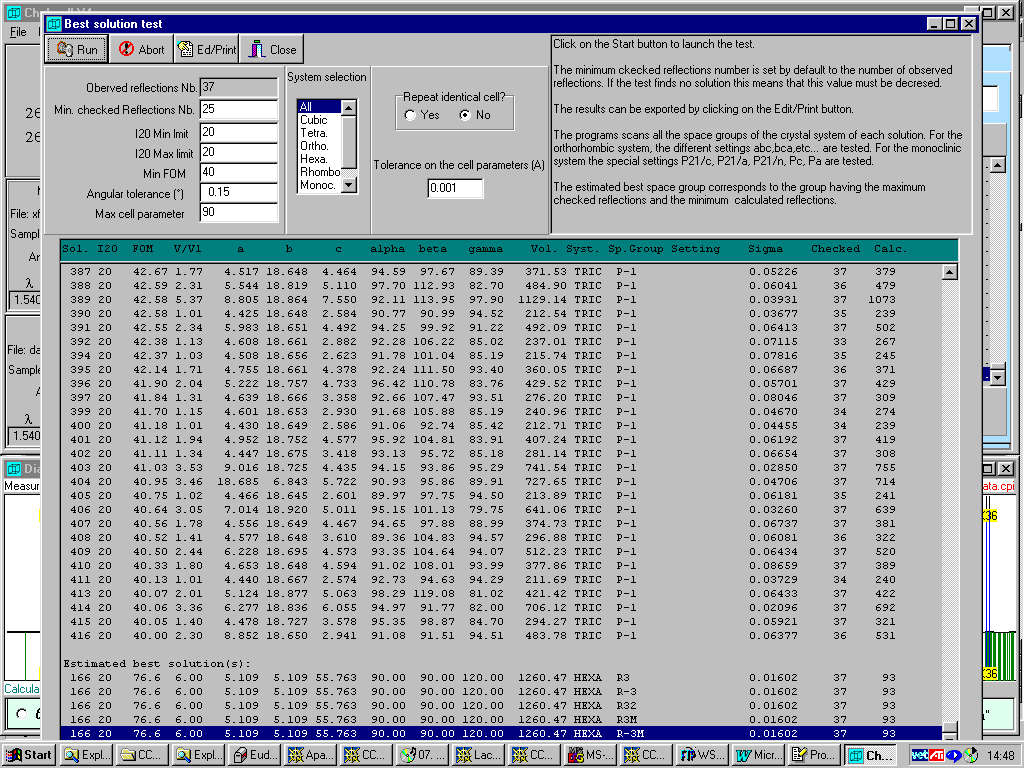
Crysfire interlinks with Chekcell for Windows (part of the LMGP suite for Windows by Jean Laugier and Bernard Bochu). Chekcell provides a graphical interface for manually and automatically suggesting a best cell/spacegroup combination using both FOM and algorithms relating to parsimony of superfluous HKLs. Latest beta version comes with Le Page and GUI Cell Transformation.
http://www.ccp14.ac.uk/tutorial/lmgp/
Chekcell has gone through the ~4000 solutions generated by Crysfire and recommended that a lower FOM (76.6) Rhombohedral (R3, R-3, R32, R-3M) cell is the best solution as found by Dicvol within Crysfire.
(this is an Inorganic Structure)NOTE: IT IS A GRAPHICAL and CONTROLLED BY A GUI!
Takes much of the
drudge work out of evaluating possible solutions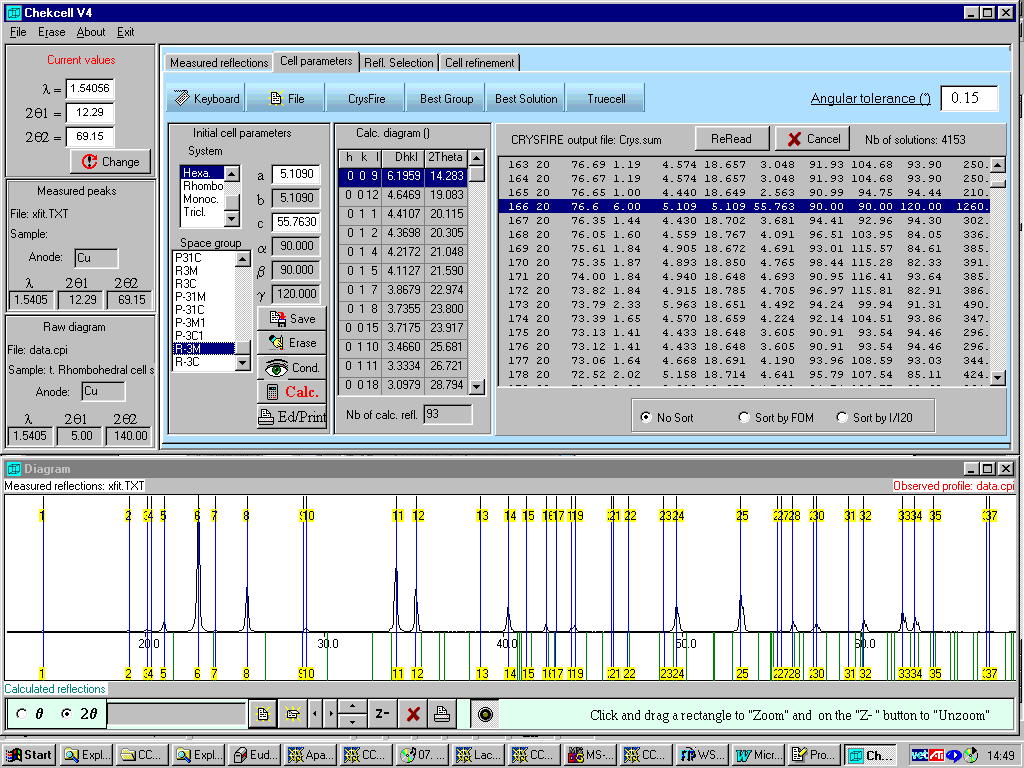
Crysfire/Chekcell:
Framework StructureUnsolvable Cell! (defined as "Treor does not solve it")
10 Minutes work for Crysfire and Chekcell to give cell and spacegroup!
ITO Solves it in this case:
1 impurity peakUnexpected Cell and Spacegroup
(though visually very compelling)
Validated after the event from a single crystal
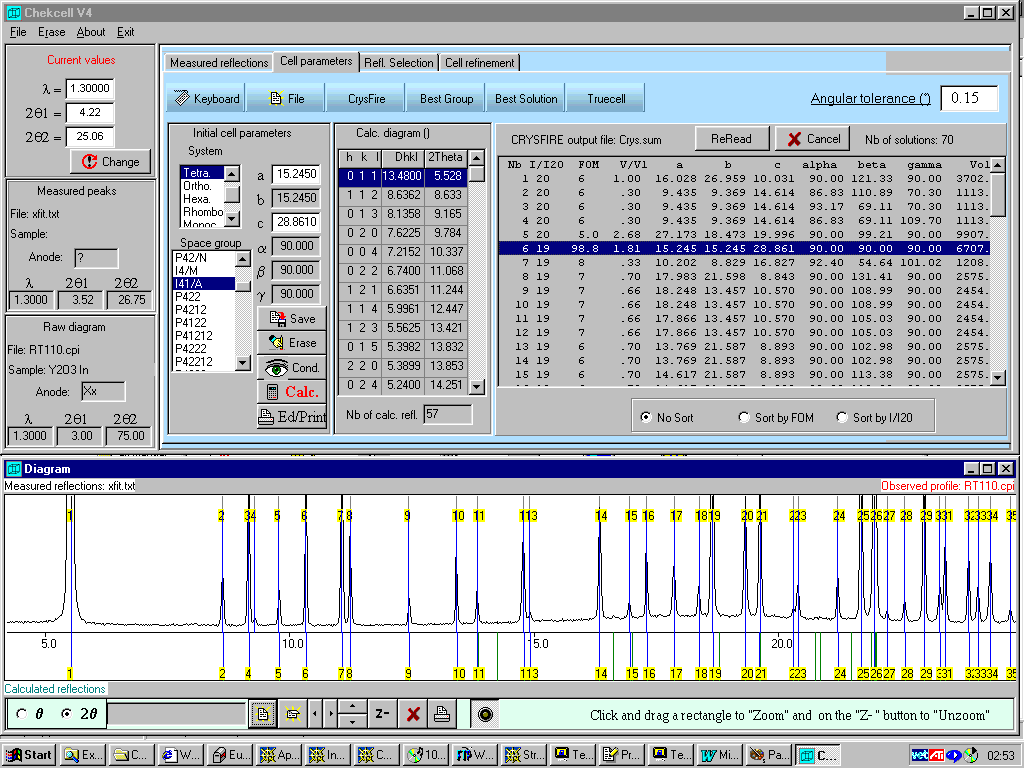
Crysfire/Chekcell for Indexing and Spacegroup Assignment
High Presssure "Angular Dispersive" Data
Kohl within Crysfire is the program that works
Structure Solved using Powder Cell menu driven Structure Transformation options
-------------------------------------------------
Very Low Resolution High Presssure
"Energy Dispersive" Data
(analogous to much laboratory organic data)
(1.5 weeks due to care in finding the best data files)
1 impurity peak on the best data files
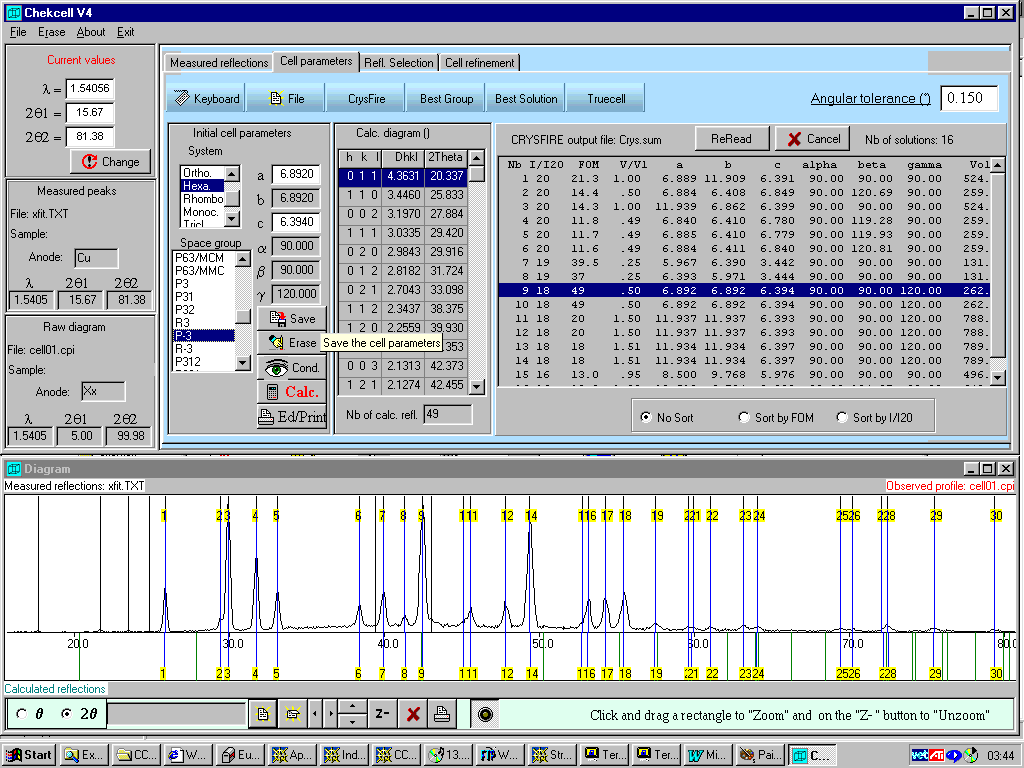
Crysfire/Chekcell for Indexing and Spacegroup Assignment
Why Isn’t this cell solving
(Organometallic)
From Armel Le Bail’s site: ESRF Powder Data as well
(very difficult problems can still be difficult on any available software program)
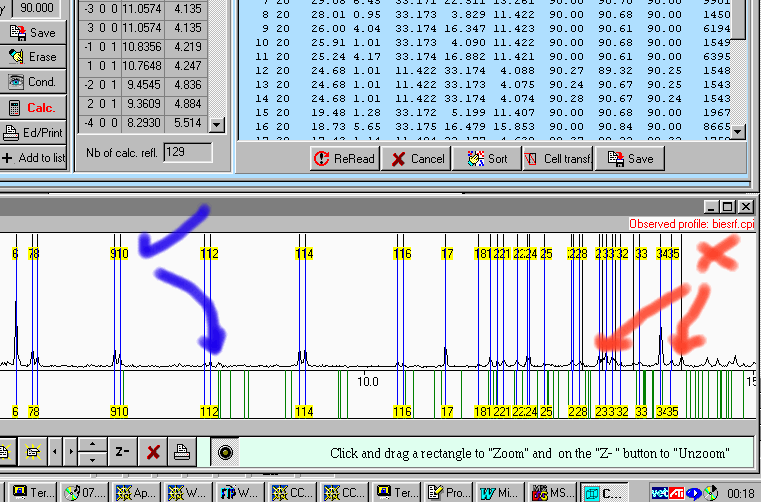
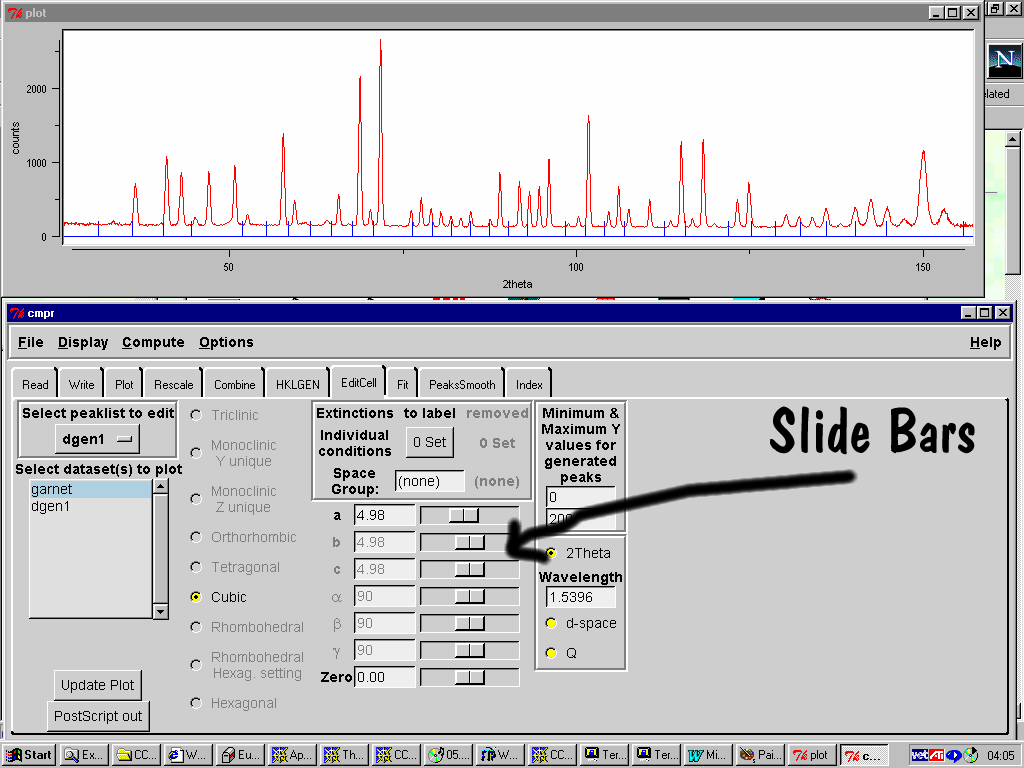
Manual Single Crystal Indexing/Spacegroup Assignment
Brian Toby’s CMPR Suite (UNIX and Windows)
(slide bars to dynamically change the cell – select spacegroup or operators)
(supercell/sub cell relationships)
http://www.ncnr.nist.gov/programs/crystallography/software/cmpr/
Single Crystal Absorption Correction Options
Ortex, Crystals, WinGX and Platon give access to a variety of Absorption correction options.
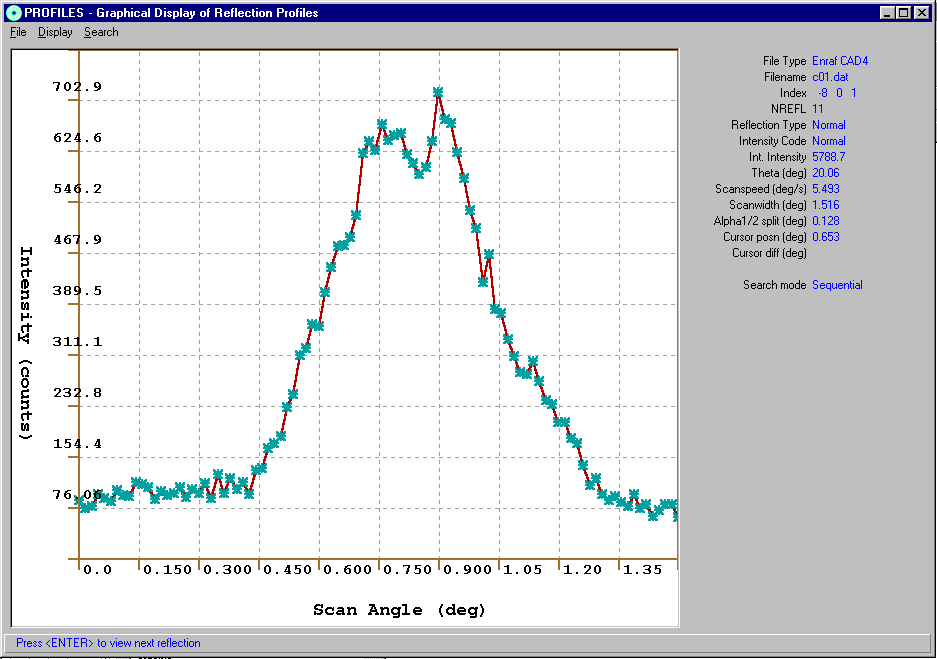
For example:
via WinGXViewing or HKL Profiles
Blessing DREAR Software
Numerical:
Gaussian, Analytical, Spherica
l, CylindricalSemi-Empirical:
Psi-scans, Camel-Jockey, Multiscan
RefDelF:
Difabs, XABS2, Shelxa
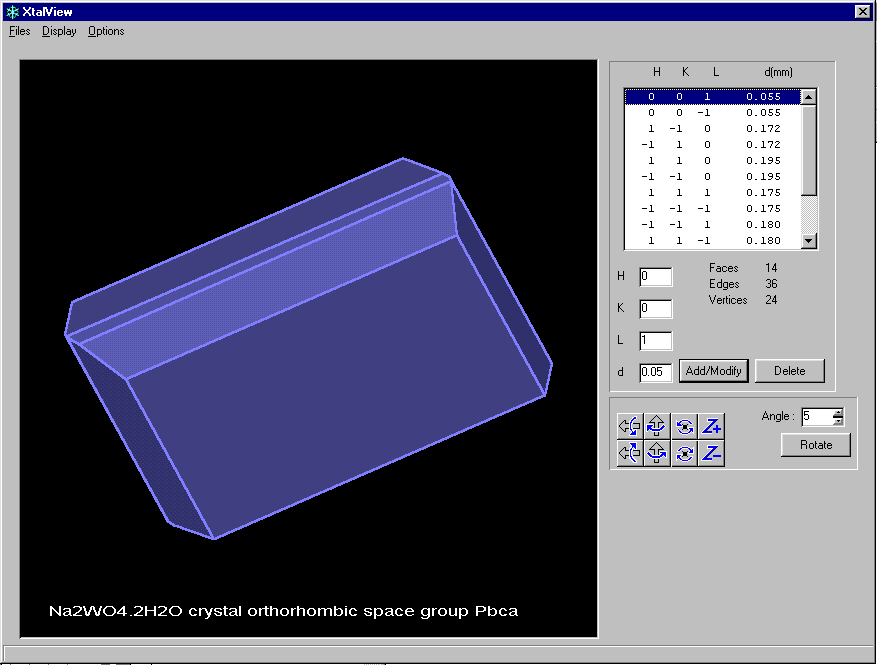
After defining faces, you can view the Crystal via
XtalView
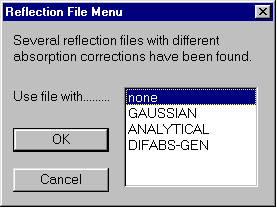
Before refinement, user is prompted which form of absorption corrected data to use:
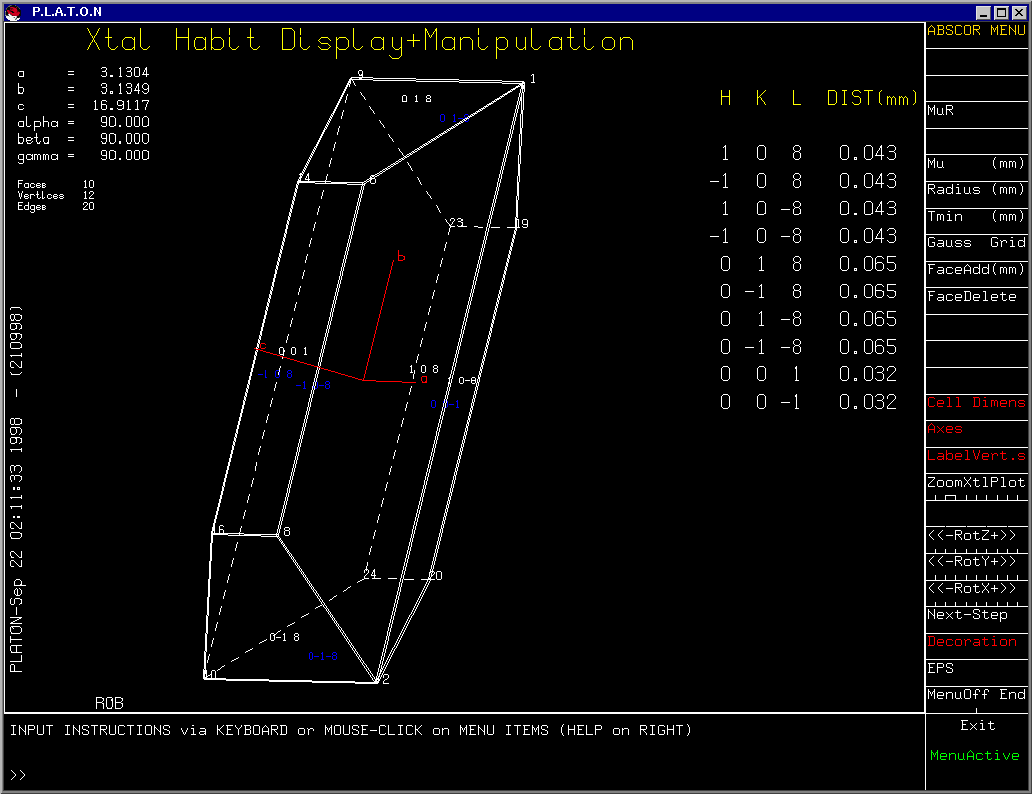
Platon
(Viewing the Crystal Habit and multiple absorption correction options)
Single Crystal Indexing/Spacegroup Assignment
(some of which is also applicable to powder diffraction)
Twinning Resources: http://www.ccp14.ac.uk/solution/twinning/
Most hardware vendors provide options for indexing but there are free-standing programs that can provide other options.
DIRAX for difficult Indexing problems.
ftp://ftp.chem.uu.nl/pub/dirax/
Twindex.
ftp://laue.chem.ncsu.edu/pub/X-ray/twindex/
The Merohedral Crystal Twinning Server
http://www.doe-mbi.ucla.edu/Services/Twinning/
The Merohedral Crystal Twinning Server
http://www.doe-mbi.ucla.edu/Services/Twinning/
TWIN3.0 for Windows (test for merohedry)
Contact V. Kahlenberg (vkahlen@uni-bremen.de)
ABSEN Single Crystal Program by Patrick McArdle
(comes with the ORTEX and WinGX suites)
http://www.nuigalway.ie/cryst/software.htm
http://www.chem.gla.ac.uk/~louis/software/
Unit Cell Refinement (Powders)
For Overall Summary of available unit cell refinement software refer to:
http://www.ccp14.ac.uk/solution/unitcellrefine/
This include:
Celref, LAPOD, Refcel, Unitcell, Eracel, Powder v2.00, XLAT, etc
Can be helpful to perform a conventional unit-cell refinement prior to a Le Bail fit (or where unit weighting of each reflection is important).
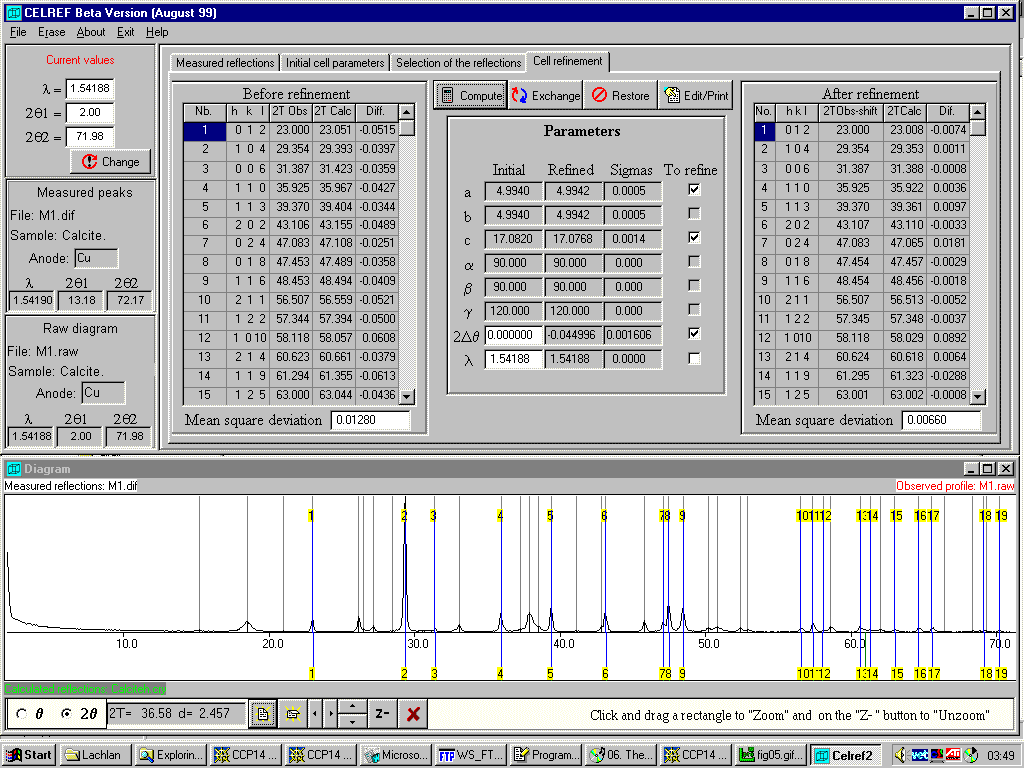
Example of Celref for Windows by Jean Laugier and Bernard Bochu performing Graphical Unit Cell refinement on calcite in a
multi-phase mixture (http://www.ccp14.ac.uk/tutorial/lmgp/) Graphics can really help sort out errors or misassigned
Full Profile Fitting (Powder)
For Overall Summary of available full profile analysis refer to:
Le Bail based: http://www.ccp14.ac.uk/solution/lebail/
Pawley Based: http://www.ccp14.ac.uk/solution/pawley/
The most common method of full profile fitting is that of Le Bail fitting: which is in most Rietveld packages. Is useful for:
Spacegroup Assignment
Unit Cell Refinement (when overlap is a problem)
Extracting Intensities for Structure Solution
Following is an example of using Fullprof Rietveld with the Winplotr for Windows program:
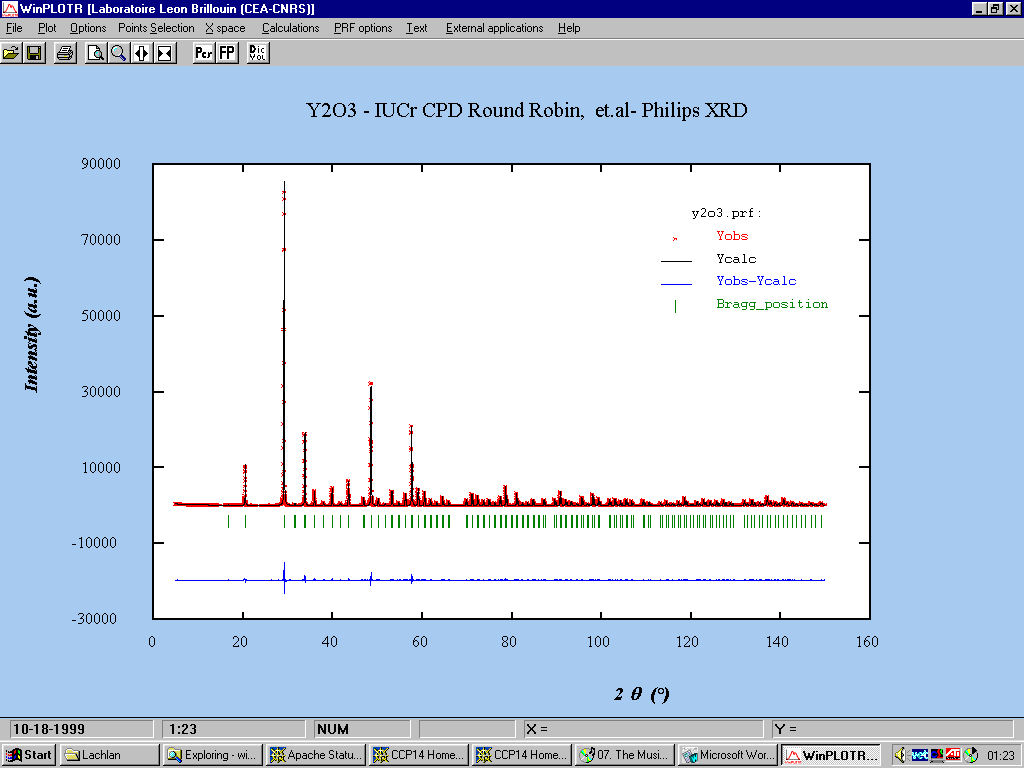
Mass unit cell refinement using Le Bail fitting
In this case using the Rietica for Windows Rietveld:
(multply the following by a few more 100 to 1000 powder patterns)
http://www.rietica.org/
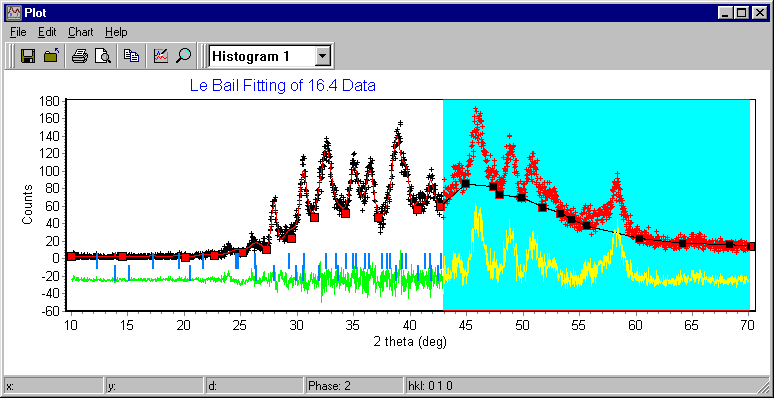
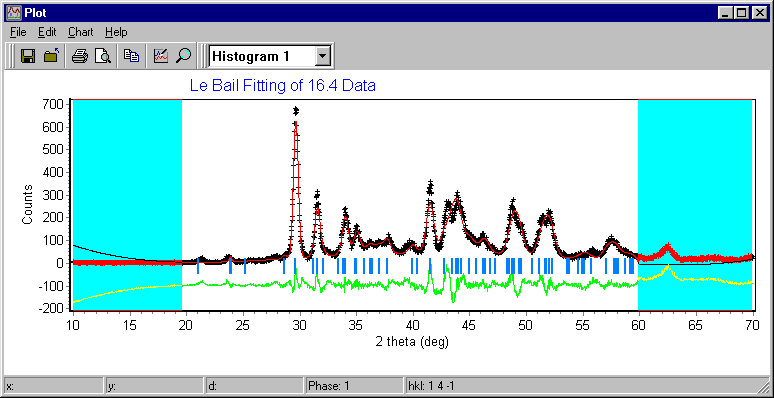
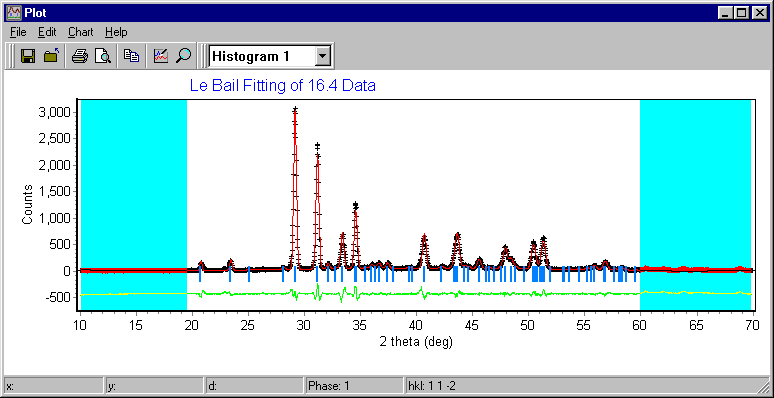
Materials Analysis Rietveld/Texture Software
Pole Figure, Texture Analysis – important also for some forms of Le Bail fitting and structure solution from powders
BEARTEX for Windows
Rudy Wenk - wenk@seismo.berkeley.edu
Texture, Pole figure, Calculates Orientation Distribution
http://www.seismo.berkeley.edu/~wenk/beartex.htm
http://www.ccp14.ac.uk/ccp/web-mirrors/beartex/~wenk/beartex.htm
GSAS for UNIX and PC
Bob Von Dreele - vondreele@lanl.gov
Menu driven interface with large vareity of functionality including Fourier map generation and viewing, ORTEP, Texture Analysis
ftp://ftp.lanl.gov/public/gsas/
http://www.ccp14.ac.uk/ccp/ccp14/ftp-mirror/gsas/public/gsas/
EXP Tcl/Tk GUI - new GUI replacement of expedit being developed by Brain Toby:
http://www.ncnr.nist.gov/programs/crystallography/
http://www.ccp14.ac.uk/ccp/web-mirrors/briantoby/programs/crystallography/
MAUD for Java
Luca Lutterotti - Luca.Lutterotti@ing.unitn.it
Sequel to Rietquan and all menu driven; concentrating on size/strain/stress analysis
http://www.ing.unitn.it/~luttero/
http://www.ccp14.ac.uk/ccp/web-mirrors/lutterotti/~luttero/
ftp://ftp.minerals.csiro.au/pub/xtallography/ccp14/ccp/web-mirrors/lutterotti/~luttero/index.html
POFINT
Daniel Chateigner - daniel.chateigner@univ-lemans.fr
Pole Figure Interpretation and Corrections
http://pecdc.univ-lemans.fr/pofint/pofint.htm
http://www.ccp14.ac.uk/ccp/web-mirrors/pofint/pofint/pofint.htm
popLA
John Bingert - bingert@lanl.gov
Texture Analysis and Modeling.
http://www.mst.lanl.gov/cms/poplalapp.html
http://www.ccp14.ac.uk/ccp/web-mirrors/popla/cms/poplalapp.html
Symmet
John Root
http://www.ccp14.ac.uk/ccp/web-mirrors/chalk_river_pole_figure/
Materials Analysis Rietveld
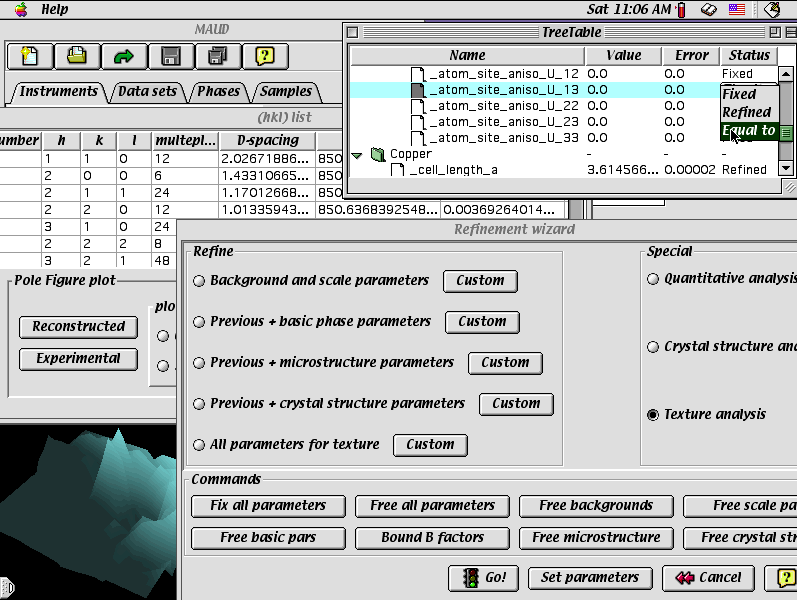
MAUD (for Java PC/Mac/UNIX)
Crystallite size and shape
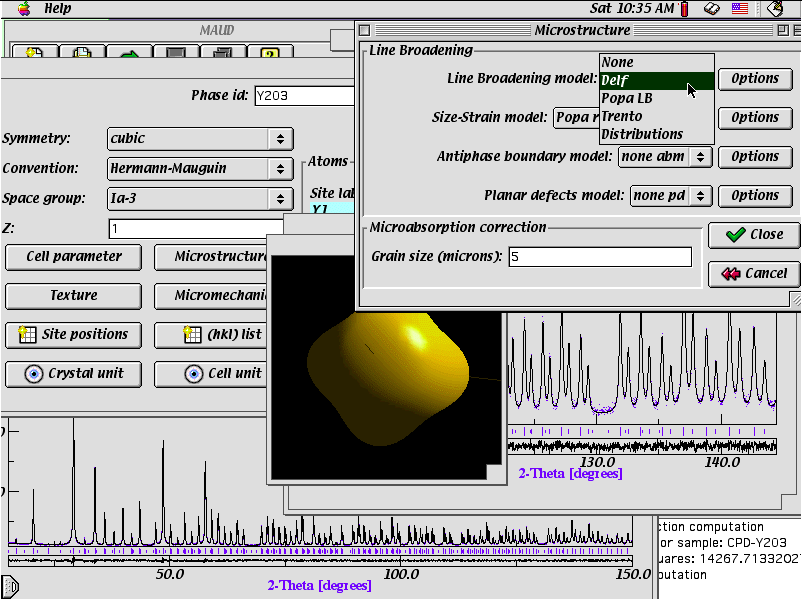
Pole figure/Refinement wizard
MAUD Quantitative Phase Analysis Tutorial:
http://www.ing.unitn.it/~luttero/maud/tutorial/
"Generic" Structure Solution from Powder Diffraction Data
Very non-trivial endeavour.
(though indexing can often be the limiting step in many attempted structure solutions)
EXPO - Direct Methods (Sirware Group) Makers of Sir92/Sir97
http://www.ba.cnr.it/IRMEC/SirWare_main.html
http://www.ccp14.ac.uk/ccp/web-mirrors/sirware/IRMEC/SirWare_main.html
If EXPO fails, used Le Bail or Pawley extracted data with Single Crystal Structure Solution Software as described below.
ESPOIR (GPL’d by Armel Le bail) Monte Carlo and pseudo simulated annealing - normally use as last resort (new version for Windows has "real space molecule and fragment location").
http://www.cristal.org/sdpd/espoir/
http://www.ccp14.ac.uk/ccp/web-mirrors/armel/sdpd/espoir/
Web tutorial on setting up the ESPOIR files in < 10 minutes" and example of solving on an organic molecule:
http://sdpd.univ-lemans.fr/sdpd/espoir/10mn/
"Specialised and Commercial" Structure Solution Programs
ZEFSA II – for Zeolites (GPL’d)
http://www.mwdeem.chemeng.ucla.edu/zefsaII/
Focus – for Zeolites
http://www.kristall.ethz.ch/LFK/software/
Fullprof – Monte Carlo for structure solution and finding Magnetic Moments in neutron data
ftp://bali.saclay.cea.fr/pub/divers/winplotr/
"Available" Commercial Structure Solution from Powder Diffraction Data software:
(Tend to have optimised algorithms and interfaces not easily available in freely obtained programs)
(Though the GPL’d ESPOIR is presently starting to match the capabilities of commercial software)
Cerius 2 by Molecular Simulations
http://www.msi.com/life/products/cerius2/
Crystal Structure Determination Package (WinCSD/CSD)
http://imr.chem.binghamton.edu/zavalij/CSD.html
DASH (Druid and Mystic of old)
http://www.ccdc.cam.ac.uk/dash/bayreuth/
DIFFRACplus TOPAS
http://www.bruker-axs.com/production/products/xrd/software/topas/
Endeavour Structure Solution from Powder Diffraction Software
http://www.crystalimpact.com/endeavour/
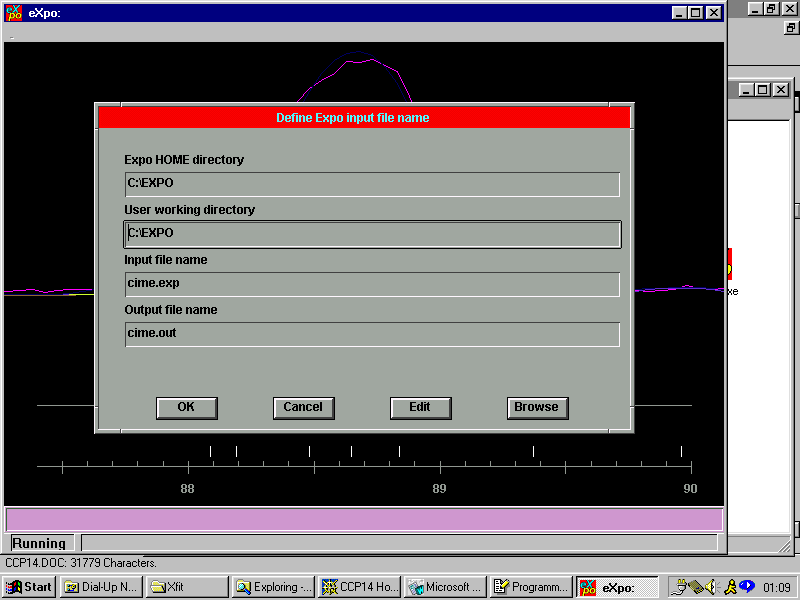
EXPO :
Direct Methods Structure Solution from Powder Data (or extracted intensities)
Initially Run with an ASCII Input File
Then Controlled by the GUI Menu
Starting Information:
%struct cime
%job cimetidine -- Synchrotron data
%init
%data
range 8.01 84.99 0.01
pattern cime.pow
cont s 4 c 40 n 24 h 64
wave 1.52904
cell 10.6986 18.8181 6.8246 90.000 111.284 90.000
space p 21/n
sync
double
%extraction
%continue
Select the Control File and Start the Solution:
Profile Fitting/ Le Bail Extraction:
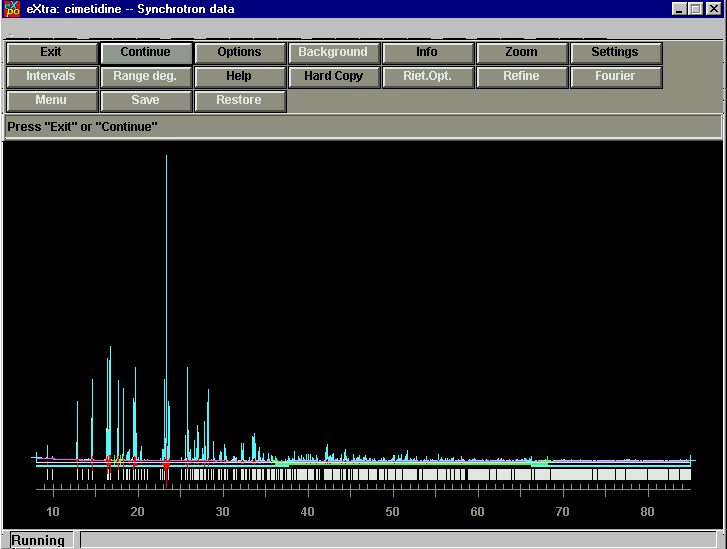
Starting the Direct Methods (at this point looks very similar to Sir 97):
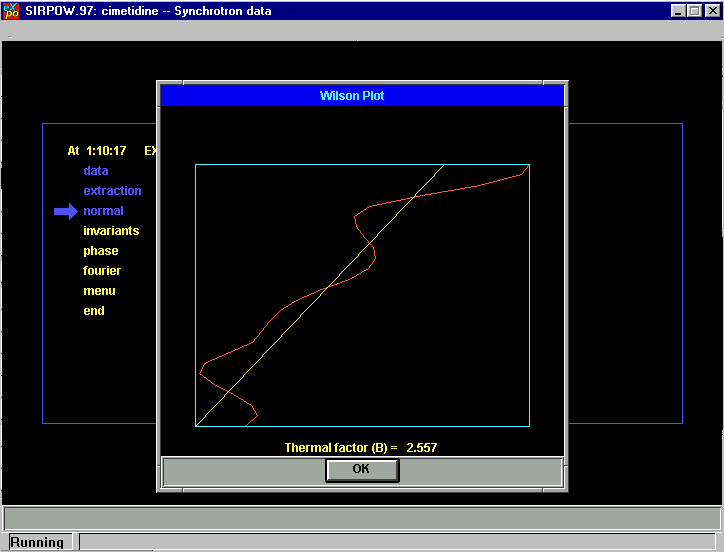
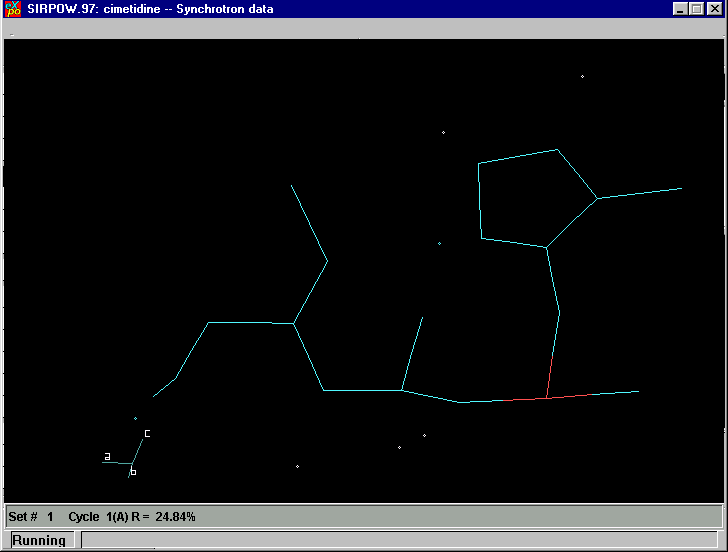
Fourier Cycling:
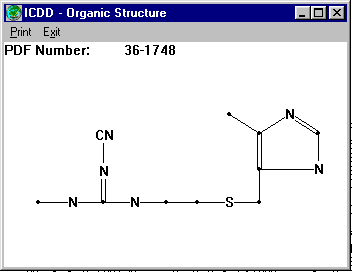
Presenting the Final Result for Evaluation (along side the idealised connectivity from the ICDD database):
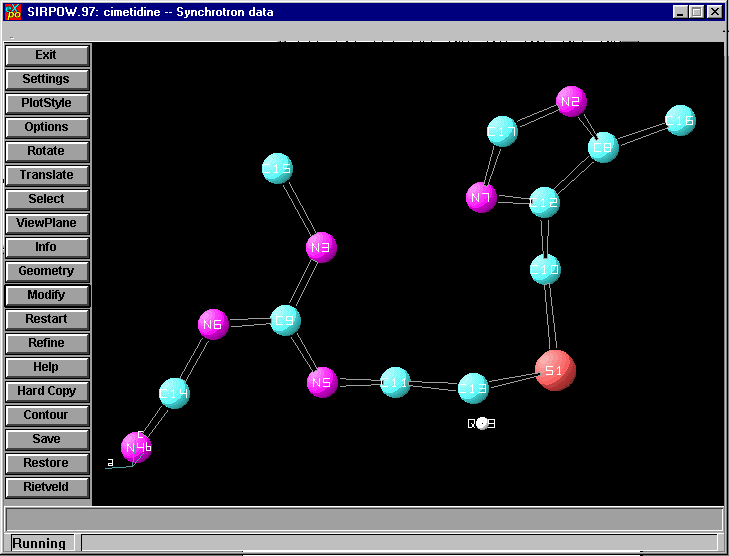
If EXPO seems to solve the structure
but looses it on the auto-building (possibly indicating a data quality problem) there are a few things that can be tried:Lower the upper limit of the powder diffraction data
RECYCLE n (placed under FOURIER, n= cycle number)
Single Crystal Structure Solution
(Applicable to extracted Powder Data)
Range of programs to choose from:
http://www.ccp14.ac.uk/solution/xtalsolution/
CAOS (also inside part of Sir97) – Ricardo Spagna, et. al.
Comes with an automatic Patterson Solution Option.
http://www.mlib.cnr.it/isc/caos/
http://www.ccp14.ac.uk/ccp/web-mirrors/caos/isc/caos/
Crisp – Part of the GPL’d Xtal Suite
Direct Methods
http://xtal.crystal.uwa.edu.au/
http://www.ccp14.ac.uk/ccp/web-mirrors/xtal/
Crunch – R. de Gelder, R.A.G. de Graaff & H. Schenk,
Direct Methods and automatic structure building
http://www-xtal.sci.kun.nl/xtal/documents/software/crunch.html
http://www.ccp14.ac.uk/ccp/web-mirrors/dirdif/xtal/documents/software/crunch.htmL
Dirdif - P.T. Beurskens, G. Beurskens, R. de Gelder, et al.
Patterson Methods for heavy atoms and fragments and automatic structure building
http://www-xtal.sci.kun.nl/xtal/documents/software/dirdif.html
http://www.ccp14.ac.uk/ccp/web-mirrors/dirdif/xtal/documents/software/dirdif.html
Windows: http://www.chem.gla.ac.uk/~louis/dirdif/
http://www.ccp14.ac.uk/ccp/web-mirrors/farrugia/~louis/dirdif/
Patsee – E. Egert and G. Sheldrick
Fragment Search
http://www.org.chemie.uni-frankfurt.de/egert/html/patsee.html
http://www.ccp14.ac.uk/ccp/web-mirrors/patsee/egert/html/patsee.html
Single Crystal Structure Solution (cont’d)
Shake’n’Bake (SnB) – Weeks, Miller, et al.
Dual-space direct methods. (Linux, SGI, IBM AIX, Alpha executables via web)
http://www.hwi.buffalo.edu/SnB/
http://www.ccp14.ac.uk/ccp/web-mirrors/snb/SnB/index.html
ShakePSD/DS*SYSTEM – Kenji Okada
Windows based direct methods for large structures up to 500 atoms
http://www.ccp14.ac.uk/ccp/web-mirrors/okada/
Shelxs 86/97/d- George Sheldrick
Direct Methods and Patterson Option
http://shelx.uni-ac.gwdg.de/SHELX/index.html
(SHELXD – for large structures. Beta versions are available from shelx ftp site )
Sir92/97/2000 – Sirware Group: Cascarano, Giacovazzo el al
Direct Methods and automatic structure building
http://www.ba.cnr.it/IRMEC/SirWare_main.html
http://www.ccp14.ac.uk/ccp/web-mirrors/sirware/IRMEC/SirWare_main.html
Solver – in NRCVAX Suite – based on Multan
Direct Methods
Peter White (pwhite@pyrite.chem.unc.edu)
XFPA – Frantisek Pavelcik
Patterson Methods and automatic structure building
E-mail: pavelcik@fns.uniba.sk
Single Crystal Examples. Summary:
1) Push the button,
2) Structure solves
3) If not, try next program (using the benefits of having access to multiple programs with different strengths) Single Crystal Suites make it trivial to easily use multiple programs.
Shelxs97
direct methods:http://shelx.uni-ac.gwdg.de/SHELX/
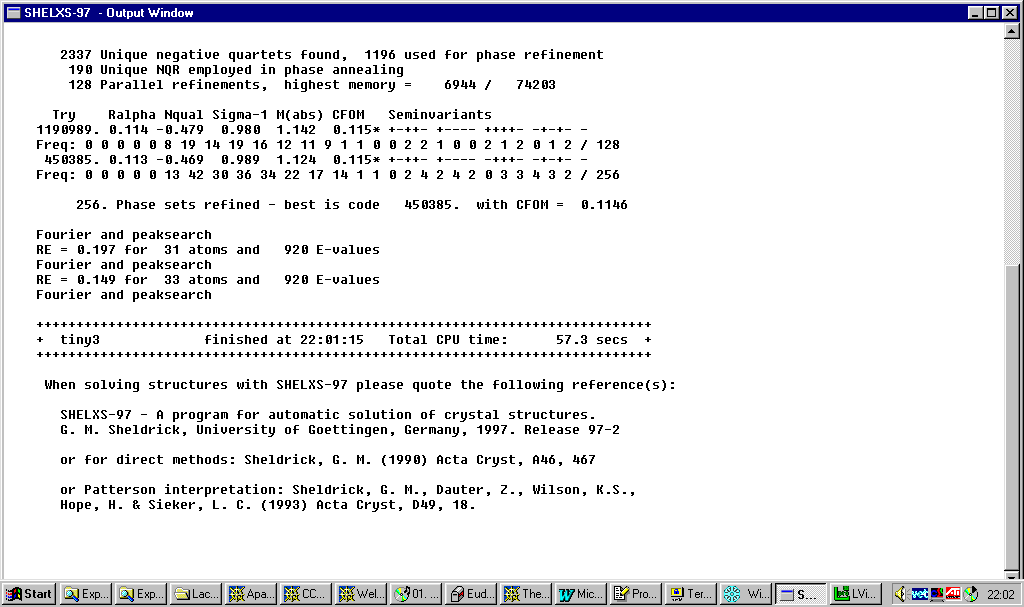
Resulting structure ready for building up:
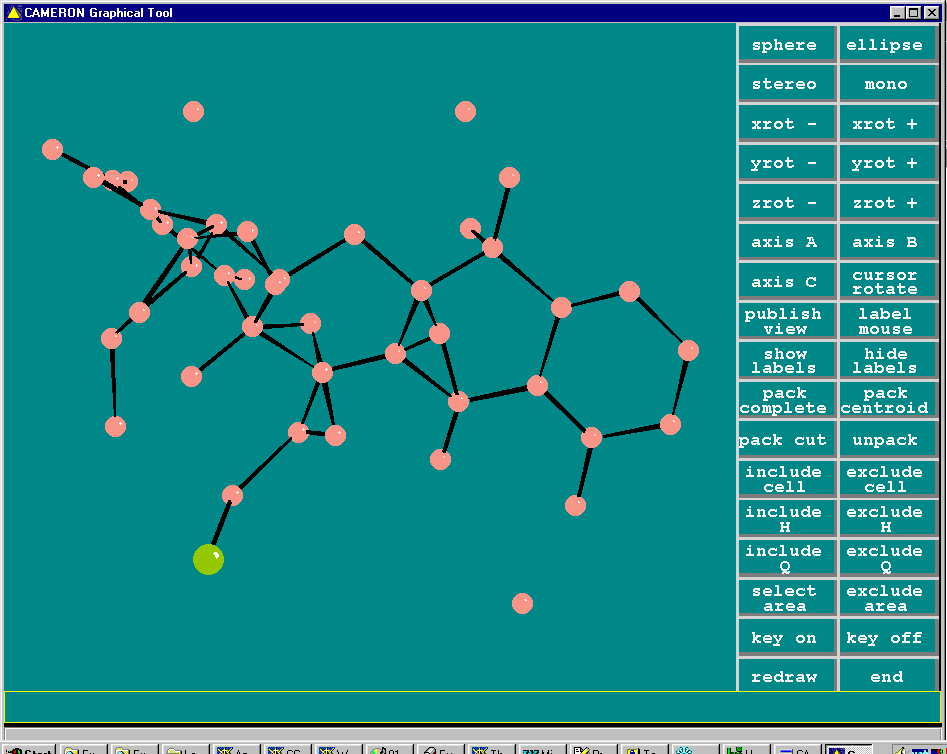
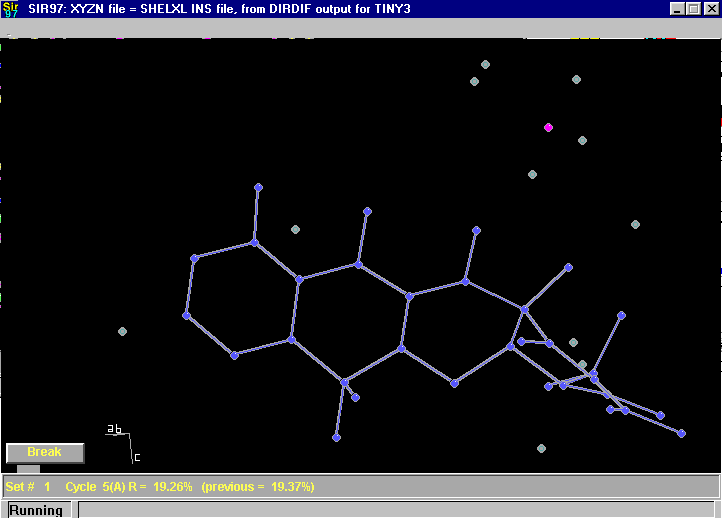
Sir97 direct methods:
http://www.ba.cnr.it/IRMEC/SirWare_main.html
http://www.ccp14.ac.uk/ccp/web-mirrors/sirware/IRMEC/SirWare_main.html
http://www.ccp14.ac.uk/tutorial/sir97/
Here Sir97 is just at the point of automatically assigning atom types during the automatic building up of a structure:
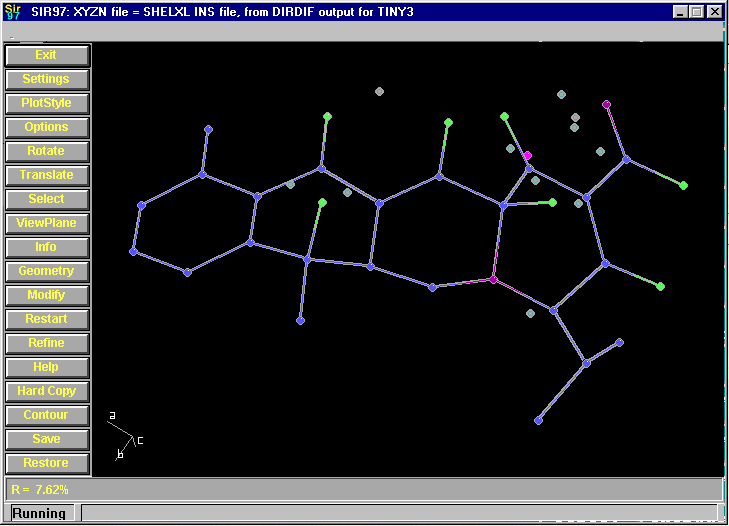
After Completion (just have to rename 3 atoms):
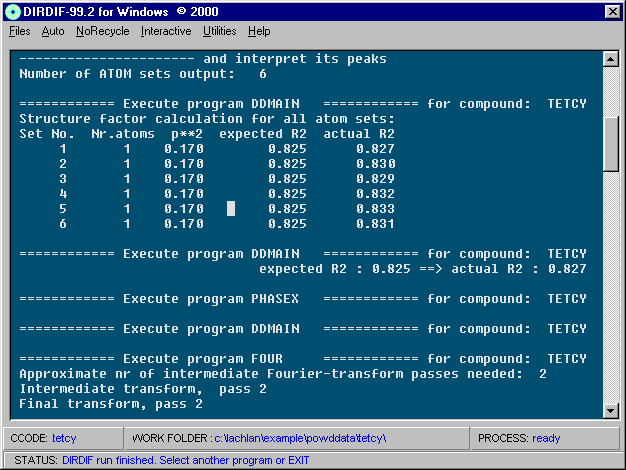
Dirdif solving a Structure. Program of choice for heavy atom problems.
http://www.ccp14.ac.uk/tutorial/dirdif/
http://www-xtal.sci.kun.nl/xtal/documents/software/dirdif.html
http://www.ccp14.ac.uk/ccp/web-mirrors/dirdif/xtal/documents/software/dirdif.html
Windows: http://www.chem.gla.ac.uk/~louis/dirdif/
http://www.ccp14.ac.uk/ccp/web-mirrors/farrugia/~louis/dirdif/
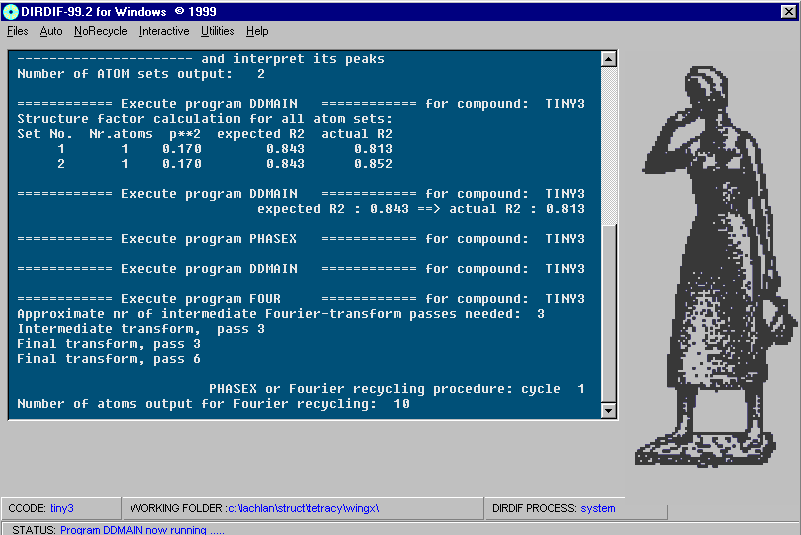
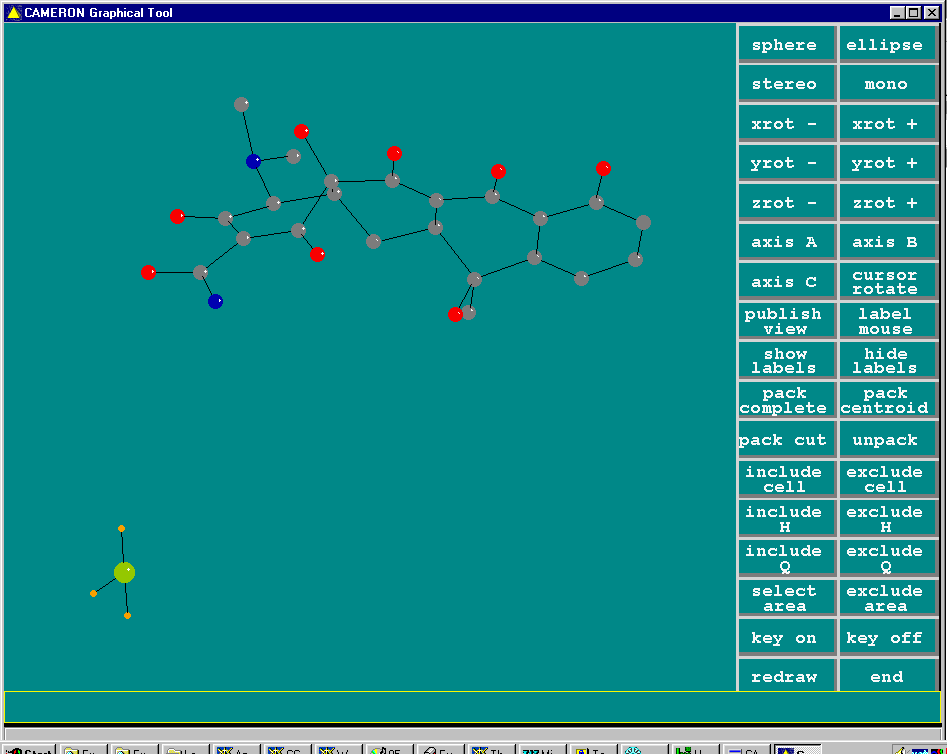
Resulting structure with every assigned non-Hydrogen atom correctly placed and named:
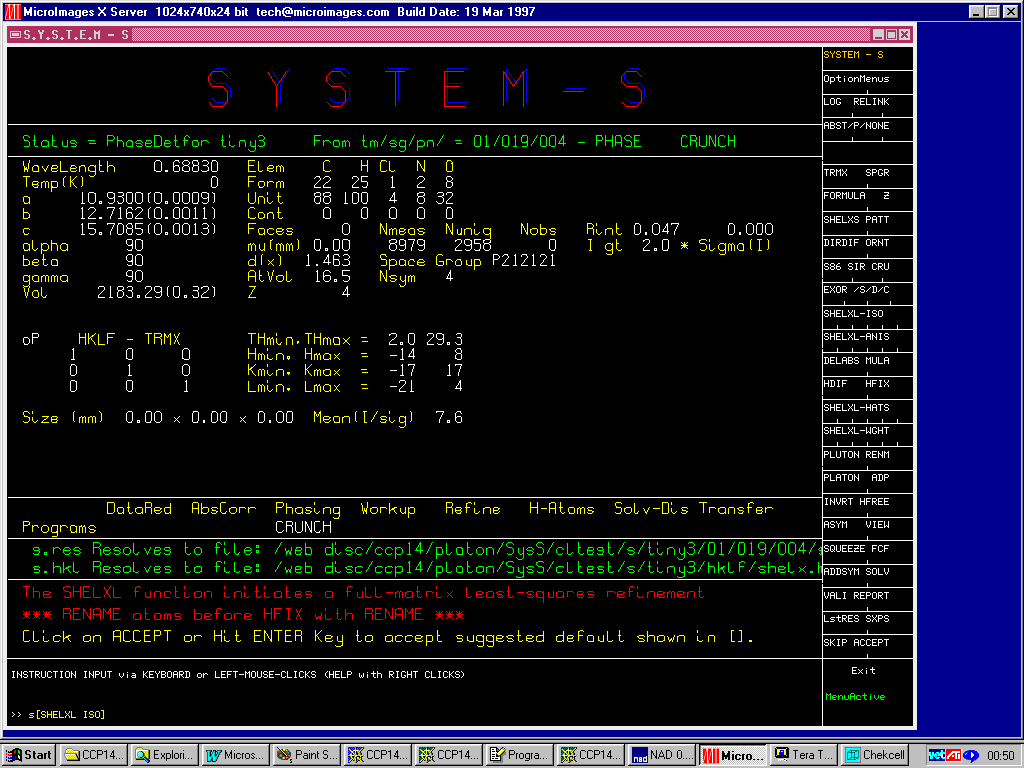
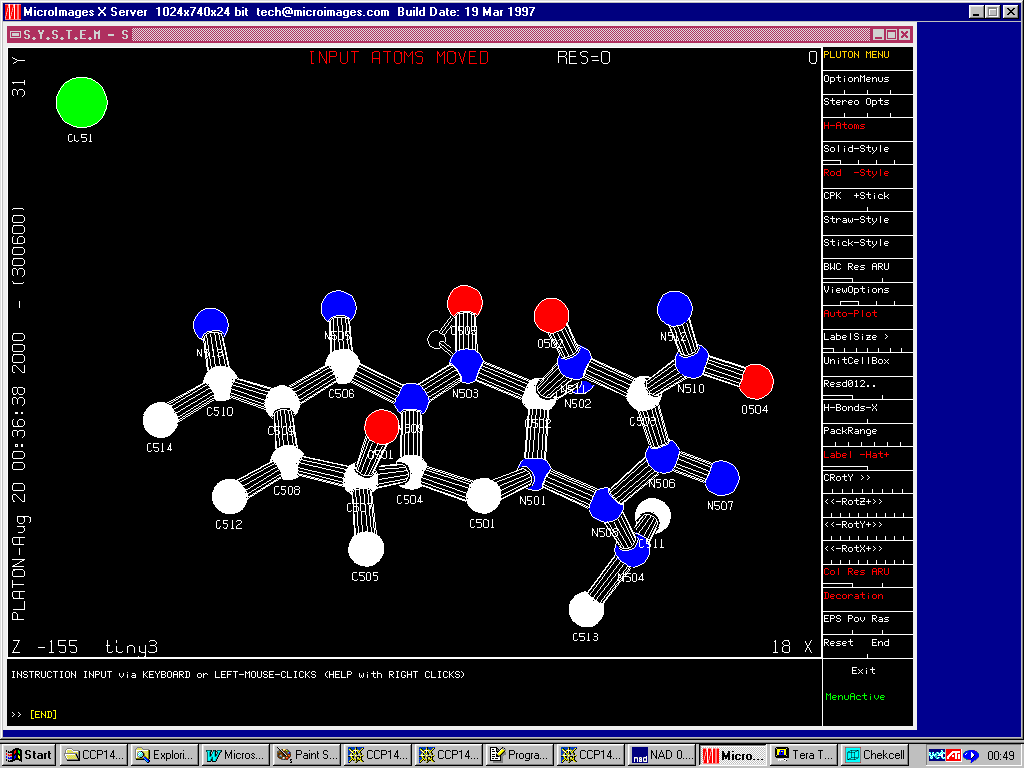
Crunch
(New December 2000 version 1.1)
http://www.ccp14.ac.uk/tutorial/crunch/
Structure solved (in this cases via the Platon/System S suite) and built to near completion (though this is really a heavy atom problem). One missing Carbon atom and various atom reassignments to be done.
Shake’n’Bake (SnB) v2.1 (large structures)
(Linux, SGI, IBM AIX, and Alpha Executables available at present)
Information and Install Tutorials for Linux:
http://www.ccp14.ac.uk/tutorial/snb/
Java GUI Interface will then take you through setting up SnB up to visualising resulting structures. Is not for those who like speedy results.
Though in a recent July 2000 "as yet unpublished" structure
SnB 2.1 gave the correct solution in around 30 minutes
(~250 atoms in the asymmetric unit). 600MHz PC / Linux
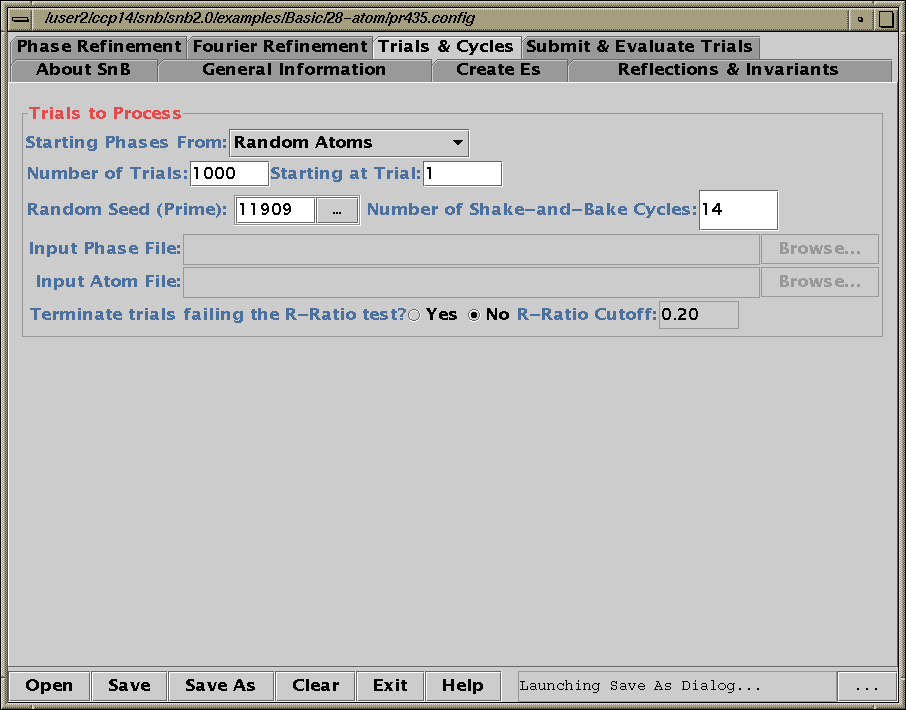
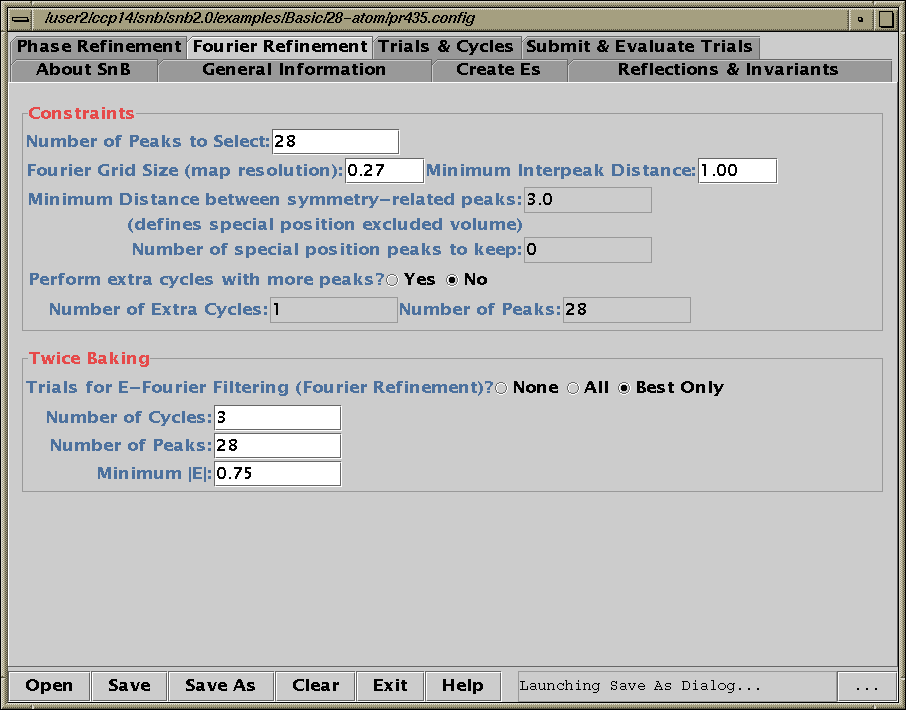
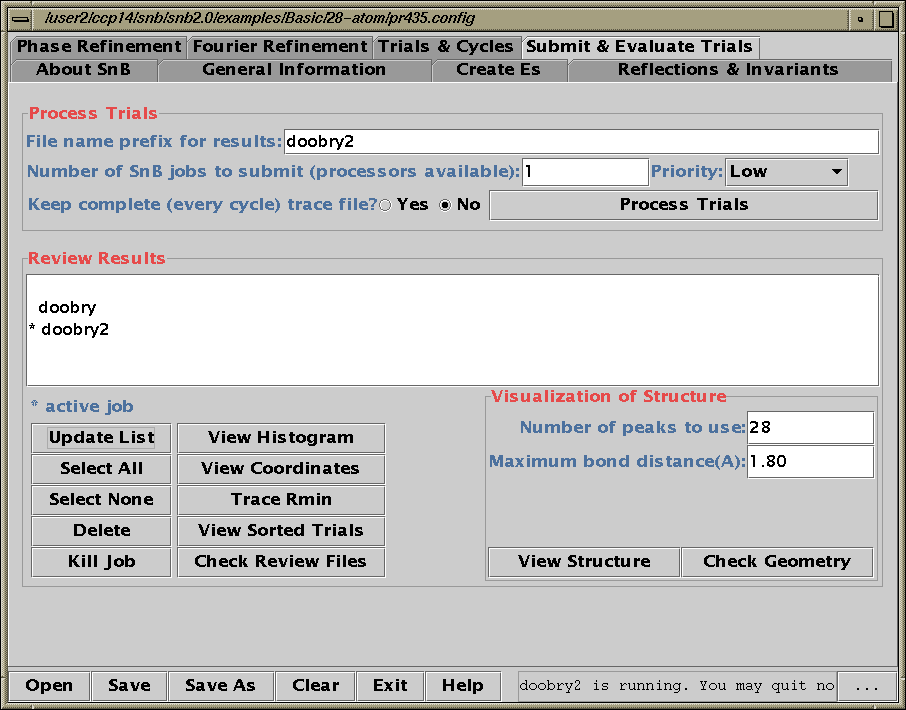
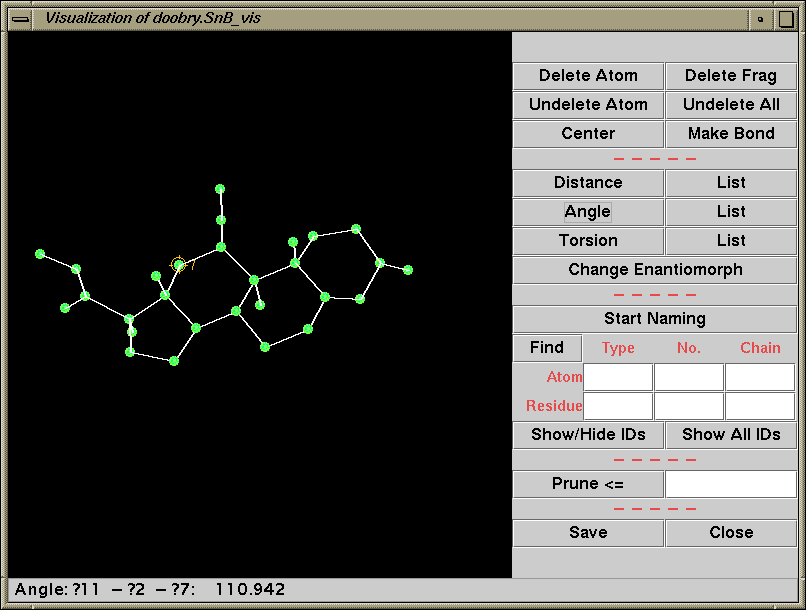
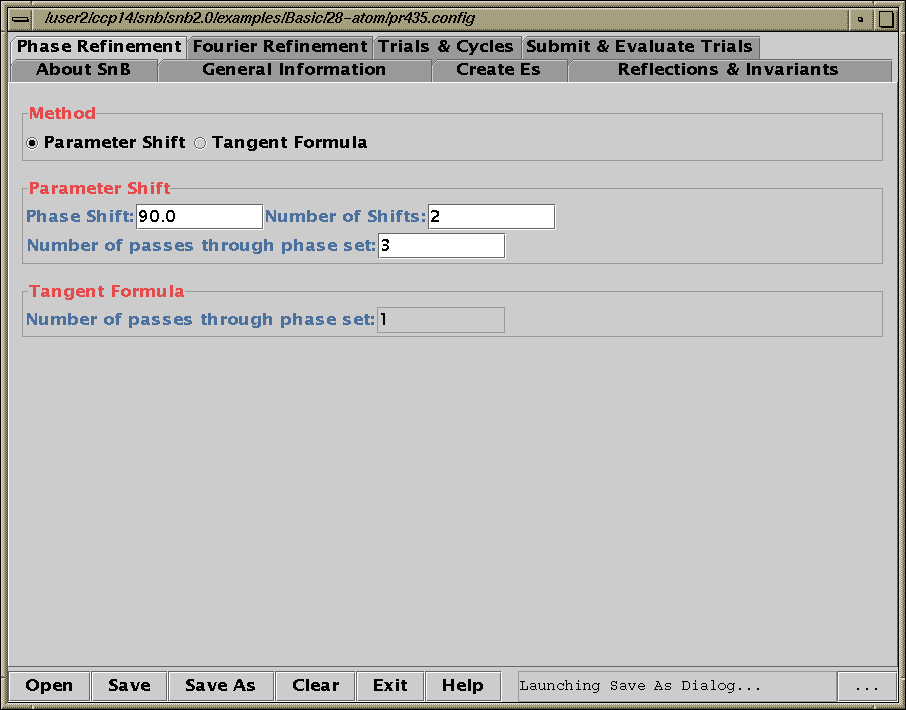
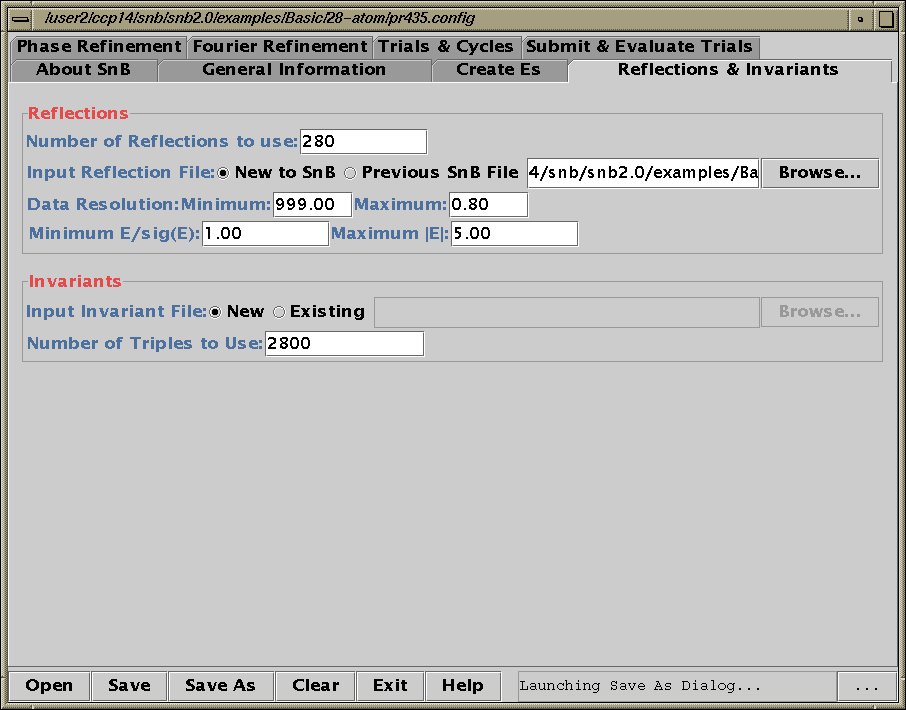
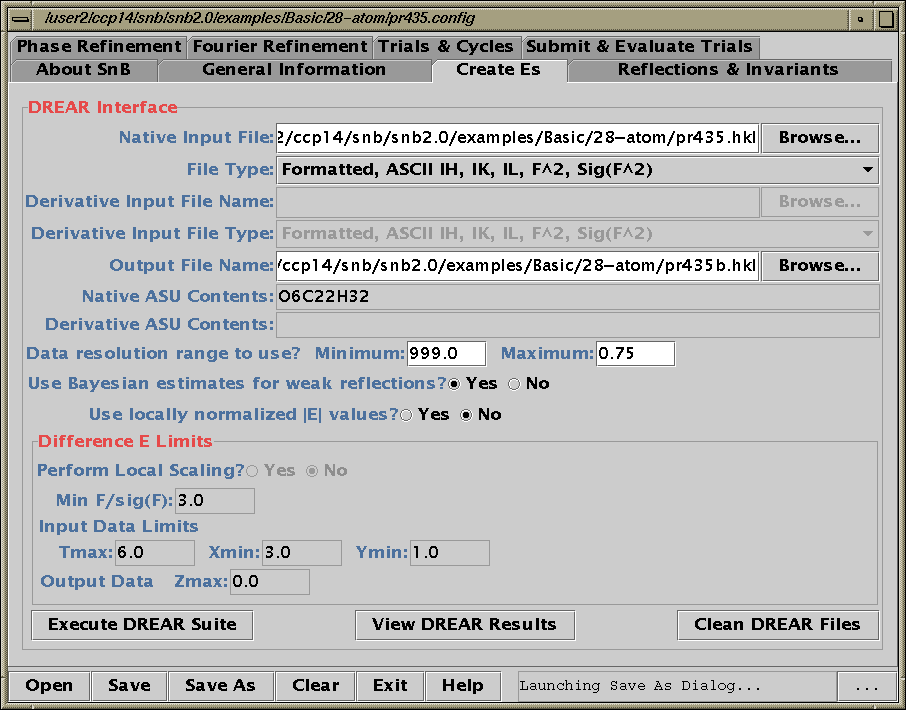

Two new packages for large structures:
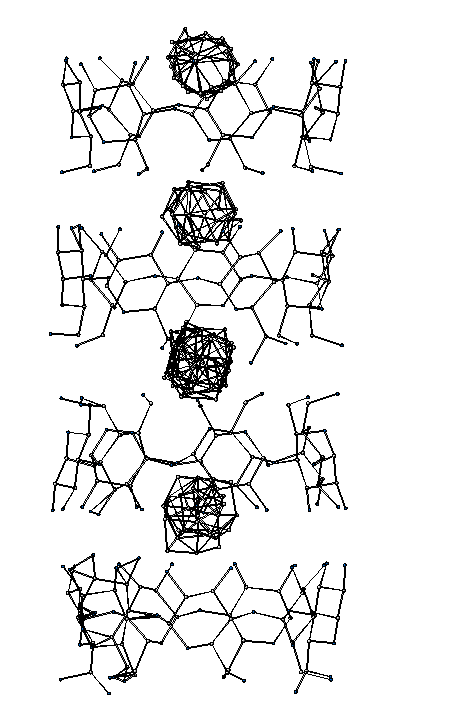
On a new Cyclodextrin structure
(Expected > 1000 atoms in the asymmetric unit). Was in the "unsolvable bin" prior to being passed on to the Sirware Group and George Sheldrick): Both Sir2000 and ShelxD programs solved the Cyclodextrin in default modes:ShelxD
9 hours on 800Mhz Athlon running LinuxSir2000
103 minutes on a Compaq XP10000(times are not directly comparable as are different computers)
2D-3D Model Building Software
(Applicable for Generating 3D fragments for Patsee/Dirdif Orient – single crystal/powder)
Refer:
http://www.ccp14.ac.uk/solution/2d_3d_model_builders/
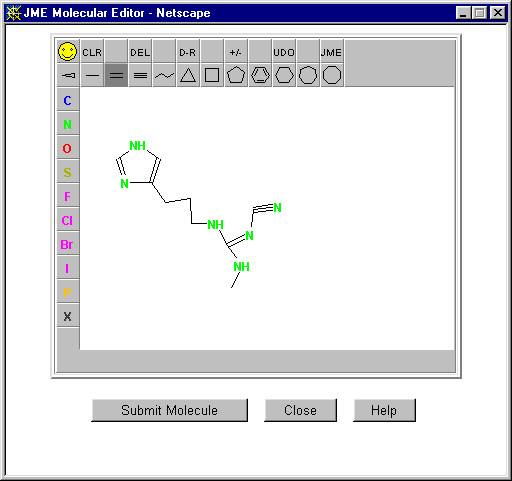
E.g., CORINA (COoRdINAtes) (Use web based direct submission):
http://www2.ccc.uni-erlangen.de/software/corina/free_struct.html
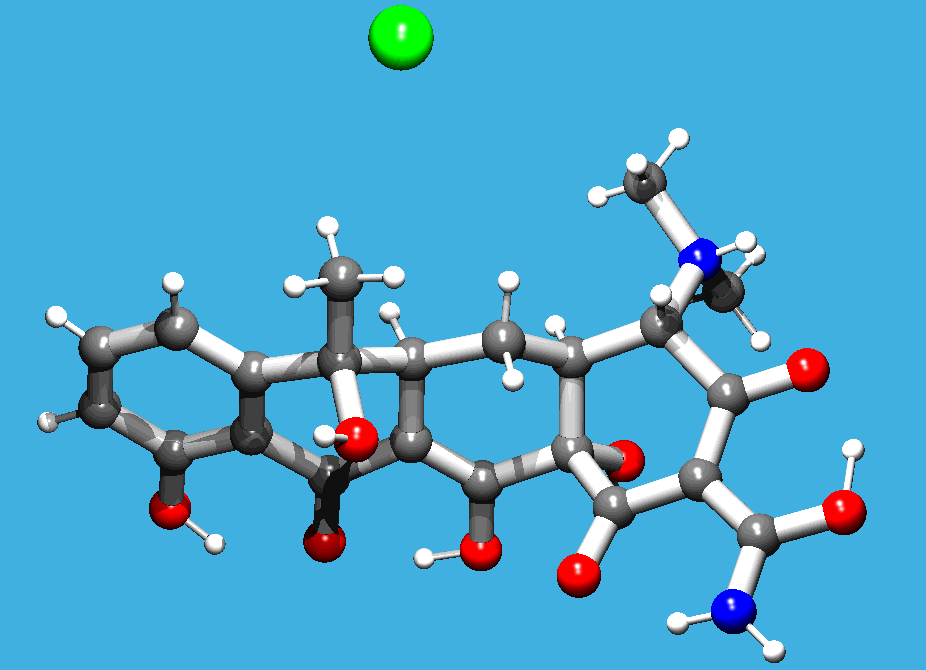
Comes with a Java Molecule Editor for building up the 2D structure over the web which generates the required SMILES string from the drawn molecule. In the following example a 2D tetracyline PDB file is generated: CN(C)C3C(O)=C(C(N)=O)C(O)C4(C)C(O)C2C(=O)c1c(O)cccc1C(C)(O)C2CC34
(Word of warning: the "energy minisation" may generate an inaccurate 3D model where different conformations are possible)
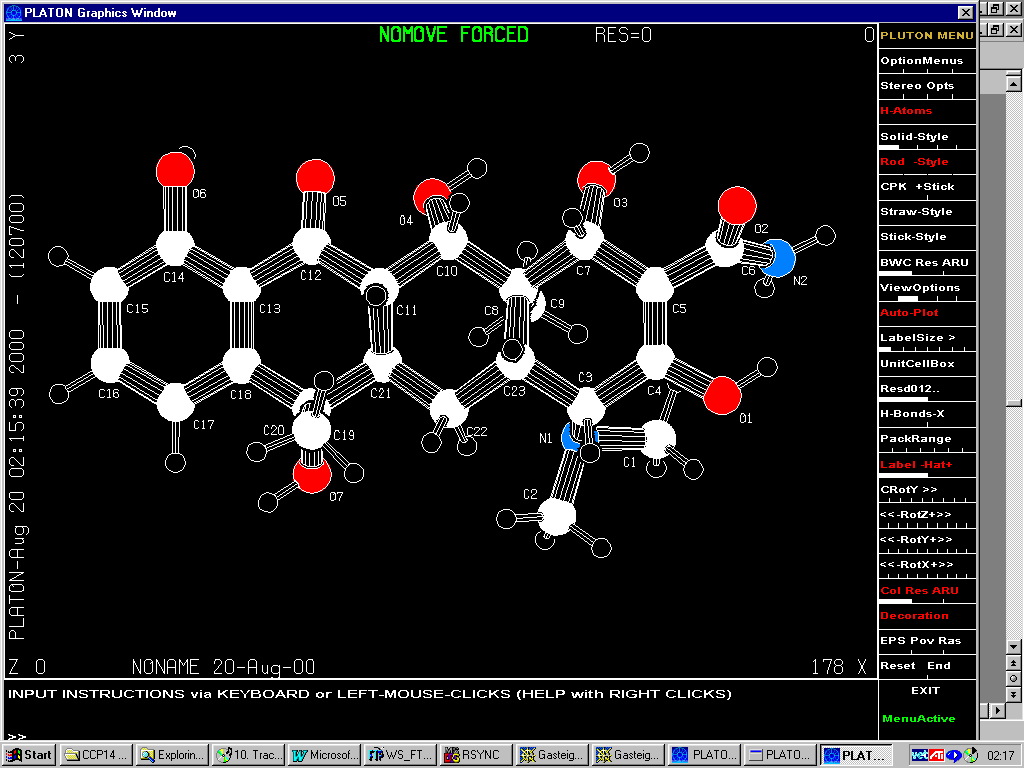
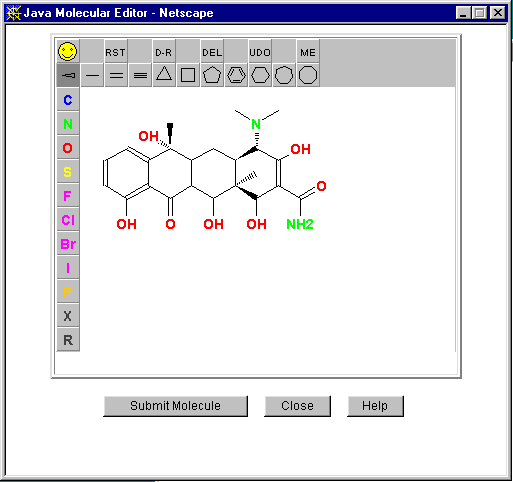
Getting Fragments intoDirdif/Dirdif for Windows.
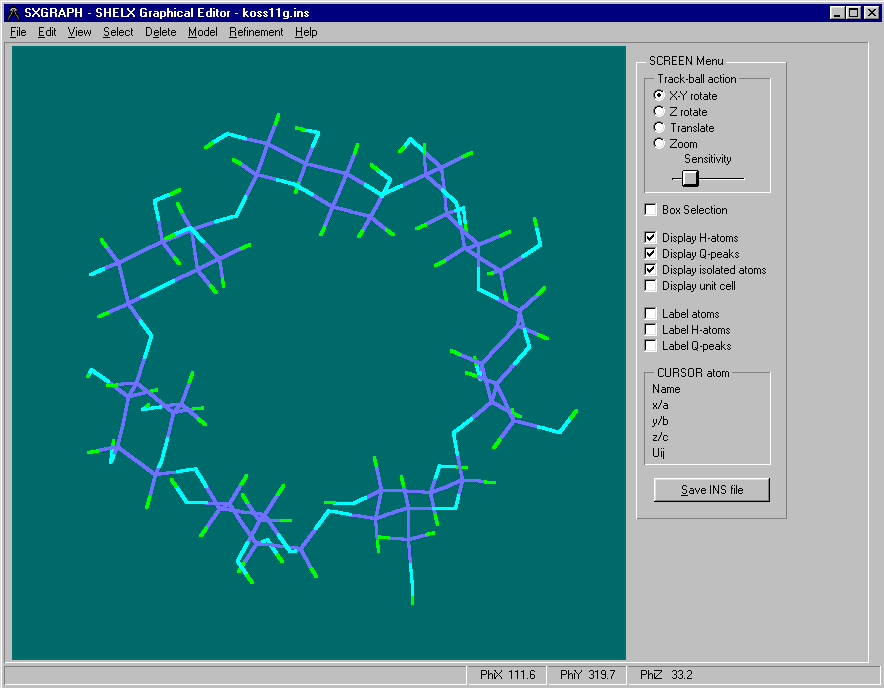
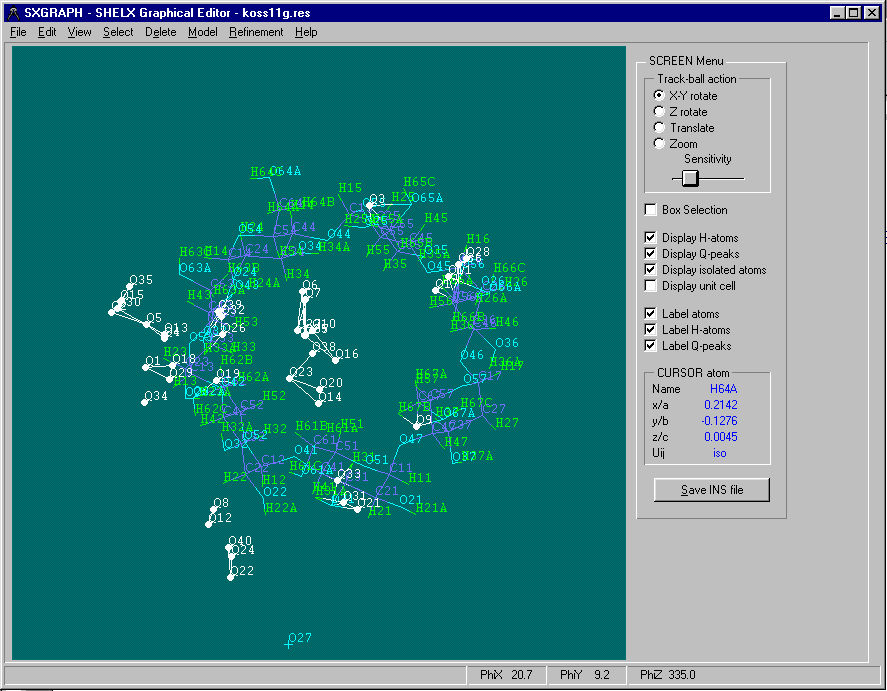
One of the User Friendliest methods is to use is WinGX’s "SXGRAPH" GUI Shelx INS/RES file Editor
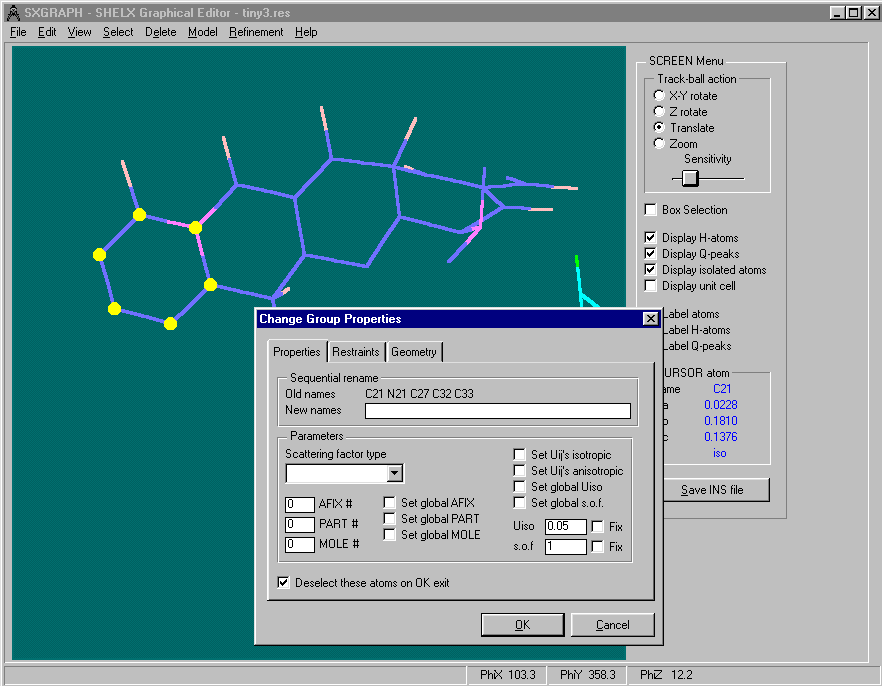 (includes ability to manipulate structures for Dirdif fragment searching)
(includes ability to manipulate structures for Dirdif fragment searching)
Either graphically Browse and Edit the Orbase Entries or Open an imported structure file (CSSR, CSD, Shelx or CIF from existing structure refinement), clean it up, then save it as a fragment ready for immediate use with Dirdif for Windows. (or any Dirdif)
(In the following case, a Cyclodextrin Fragment)
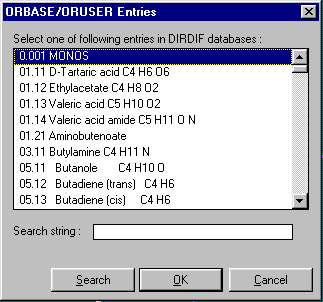
Single Crystal Structure Refinement
(Applicable to powder diffraction for helping build up the structure)
Range of programs to choose from:
http://www.ccp14.ac.uk/solution/xtalrefine/
CAOS (freestanding and also available inside Sir97)
http://www.isc.mlib.cnr.it/caos/
http://www.ccp14.ac.uk/ccp/web-mirrors/caos/caos/
http://www.ba.cnr.it/IRMEC/SirWare_main.html
http://www.ccp14.ac.uk/ccp/web-mirrors/sirware/IRMEC/SirWare_main.html
Crystals
http://www.xtl.ox.ac.uk/
http://www.ccp14.ac.uk/ccp/ccp14/ftp-mirror/crystals/pub/crystals/
DS*SYSTEM/LSBF Refinement
http://www.ccp14.ac.uk/ccp/web-mirrors/okada/
NRCVAX
Contact: pwhite@pyrite.chem.unc.edu
Shelxl (refinement program for System S/ORTEX/WinGX)
http://shelx.uni-ac.gwdg.de/SHELX/index.html
The following freely available single crystal suites are based around Shelx for Refinement:
Platon/System S http://www.cryst.chem.uu.nl/platon/
ORTEX http://www.nuigalway.ie/cryst/software.htm
WinGX http://www.chem.gla.ac.uk/~louis/software/
Xtal (GPL’d)
http://xtal.crystal.uwa.edu.au/
http://www.ccp14.ac.uk/ccp/web-mirrors/xtal/
Single Crystal Structure Refinement:
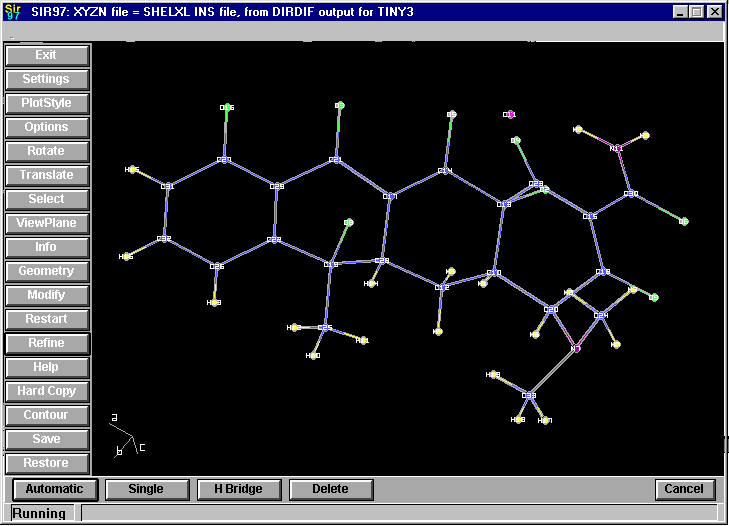
Example of Sir97/CAOS:
By default, CAOS is used via a powerful scripting language. Is inside Sir97 (point and click mode) - thus the following screen dumps are from Sir97
Adding Hydrogens
Automatic Addition of Carbon and Nitrogen bonded hydrogens at the click of a button:
Single Crystal Structure Refinement:
GUI CRYSTALS for Windows:
Adding Hydrogens
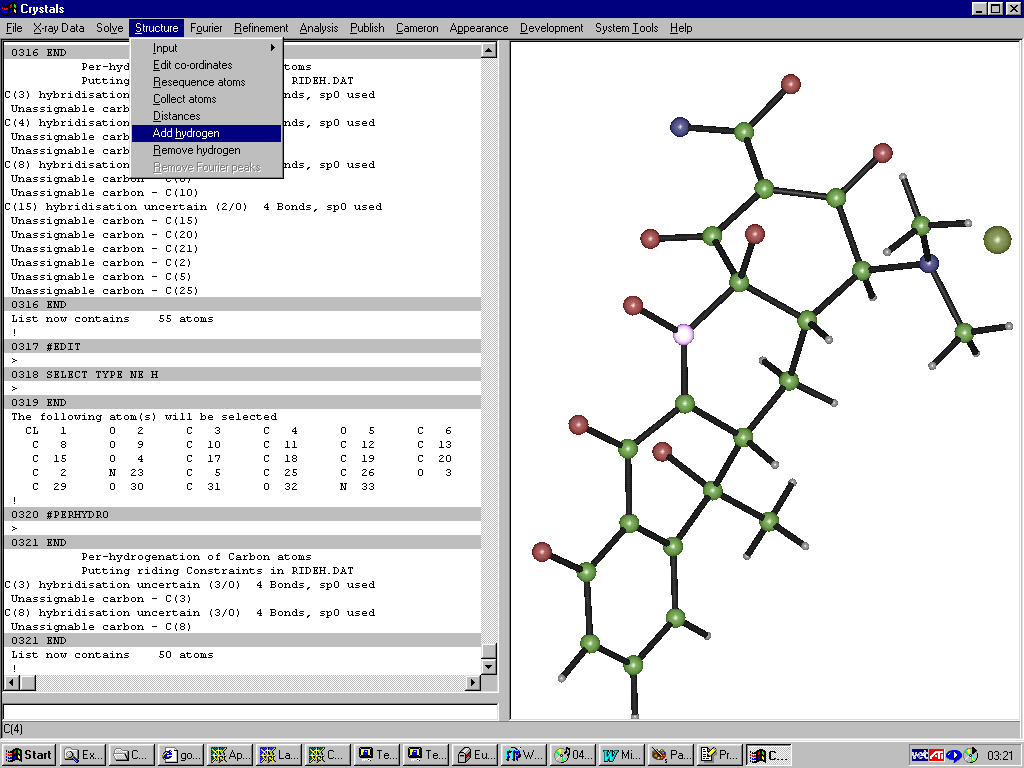
Single Crystal Structure Refinement:
Controlling a Shelx refinement via WINGX using the SXGRAPH GUI for Windows:
Adding Hydrogens
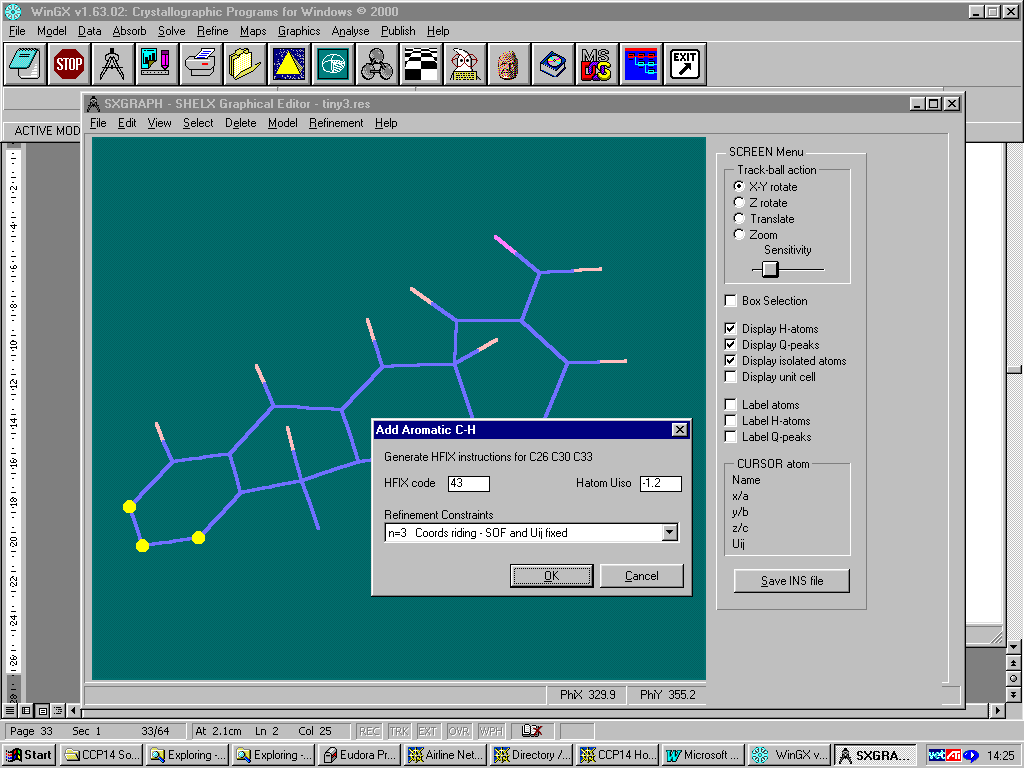
Single Crystal Structure Refinement:
CRYSTALS for Windows:
Creating and Refining Rigid Bodies
(all done in Crystallographic space)
(The "Crystals" code has been redone in "CAOS"(?))
The following is peformed using the Crystals script interface. (The new version will be full GUI based as well as traditional scripts)
Convert 5 atoms into a regularized 5 membered ring
#regu replace
group 5
{name or point and click on atoms}cp_ring <defines and regularized the rigid body>
end
Refine selected atoms as a rigid body.
#list 12
group
{name or point and click on atoms}end
Structure Refinement using Powder Diffraction Data
(Rietveld Refinement)
Large range of programs to choose from:
http://www.ccp14.ac.uk/mirror/mirror.htm
Freely Available Rietveld Software
(Contact Names are provided)
ARITVE
Armel Le Bail - armel@fluo.univ-lemans.fr
Refinement of Glasses, ASCII Control file
http://fluo.univ-lemans.fr:8001/aritve.html
http://www.cristal.org/aritve.html
http://www.ccp14.ac.uk/ccp/web-mirrors/armel/aritve.html
ftp://ftp.minerals.csiro.au/pub/xtallography/ccp14/ccp/web-mirrors/armel/aritve.html
BGMN
Joerg Bergmann - support@bgmn.de
Fundamental Parameters Rietveld
http://www.bgmn.de
http://www.ccp14.ac.uk/ccp/web-mirrors/bgmn/
ftp://ftp.minerals.csiro.au/pub/xtallography/ccp14/ccp/web-mirrors/bgmn/index.html
DBWS
Ray Young - r.young@physics.gatech.edu
DBWS Style Control File, Includes a DB2dI program for converting a Rietveld output into an appropriate D/I format suitable for submission to the ICDD.
Contact the author on the above E-mail address.
DEBVIN
Sergio Bruckner - sergio.bruckner@dstc.uniud.it
Rietveld refinement with generalized coordinates subjected to geometrical restraints. Applications in refinement of polymers.
ftp://ftp.cc.uniud.it/DEBVIN/
http://www.ccp14.ac.uk/ccp/ccp14/ftp-mirror/debvin/DEBVIN/
ftp://ftp.minerals.csiro.au/pub/xtallography/ccp14/ccp/ccp14/ftp-mirror/debvin/DEBVIN/
EXPO
Sirware Group - cryst@area.ba.cnr.it
While mainly for Structure Solution, also has menu driven Rietveld.
http://www.ba.cnr.it/IRMEC/SirWare_main.html
http://www.ccp14.ac.uk/ccp/web-mirrors/sirware/IRMEC/SirWare_main.html
Fullprof for UNIX and PC
Juan Rodriguez-Carvajal - juan@bali.saclay.cea.fr
DBWS Style ASCII Control file with large variety of functionality.
ftp://charybde.saclay.cea.fr/pub/divers/
http://www.ccp14.ac.uk/ccp/ccp14/ftp-mirror/fullprof/pub/divers/
ftp://ftp.minerals.csiro.au/pub/xtallography/ccp14/ccp/ccp14/ftp-mirror/fullprof/pub/divers/
WinPlotr Graphics link into Fullprof with enhanced functionality:
http://www-llb.cea.fr/winplotr/winplotr.htm
http://www.ccp14.ac.uk/ccp/web-mirrors/plotr/winplotr/winplotr.htm
GSAS for UNIX and PC
Bob Von Dreele - vondreele@lanl.gov
Menu driven interface with large vareity of functionality including Fourier map generation and viewing, ORTEP, Texture Analysis
ftp://ftp.lanl.gov/public/gsas/
http://www.ccp14.ac.uk/ccp/ccp14/ftp-mirror/gsas/public/gsas/
EXP Tcl/Tk GUI - new GUI replacement of expedit being developed by Brain Toby:
http://www.ncnr.nist.gov/programs/crystallography/software/expgui/expgui_intro.html
http://www.ccp14.ac.uk/ccp/web-mirrors/briantoby/programs/crystallography/software/expgui/expgui_intro.html
Koalariet
Alan Coelho - CCP14 Contact - L. Cranswick (l.cranswick@dl.ac.uk)
Fundamental Parameters Rietveld - sequel is called "TOPAS" by Bruker
http://www.ccp14.ac.uk/tutorial/xfit-95/xfit.htm
LHPM/Rietica for Win95/NT
Brett Hunter - bah@ansto.gov.au
Full GUI and/or DBWS ASCII file driven, Fourier Maps, I.D. Brown bond-valence analysis.
ftp://ftp.ansto.gov.au/pub/physics/neutron/rietveld/Rietica_LHPM95/
http://www.ccp14.ac.uk/ccp/ccp14/ftp-mirror/ansto/pub/physics/neutron/rietveld/Rietica_LHPM95/
MAUD for Java
Luca Lutterotti - Luca.Lutterotti@ing.unitn.it
Sequel to Rietquan and all menu driven; concentrating on size/strain/stress analysis
http://www.ing.unitn.it/~luttero/
http://www.ccp14.ac.uk/ccp/web-mirrors/lutterotti/~luttero/
ftp://ftp.minerals.csiro.au/pub/xtallography/ccp14/ccp/web-mirrors/lutterotti/~luttero/index.html
PREMOS/REMOS
Akiji Yamamoto - yamamoto@nirim.go.jp
Modulated Structure Refinement
http://www.nirim.go.jp/~yamamoto/
http://www.ccp14.ac.uk/ccp/web-mirrors/remos/~yamamoto/
ProDD
Jon Wright - jpw22@cam.ac.uk
Based on the CCSL profile refinement routines
http://www.cus.cam.ac.uk/~jpw22/
Profil
Jeremy Cockcroft - cockroft@gordon.cryst.bbk.ac.uk
Part of the PDPL Powder Diffraction Suite
ftp://img.cryst.bbk.ac.uk/pdpl/
http://www.ccp14.ac.uk/ccp/ccp14/ftp-mirror/profil/PDPL/
Riet7/SR5
Ian Madsen - ian.madsen@minerals.csiro.au
DBWS Style Control File, Variable Count Time Data
ftp://ftp.minerals.csiro.au/pub/xtallography/
http://www.ccp14.ac.uk/ccp/ccp14/ftp-mirror/csirominerals-anon-ftp/pub/xtallography/
RIETAN-2000 (GPL’d)
Fujio Izumi - izumi@nirim.go.jp
Whole-pattern fitting based on the maximum-entropy method (MEM)
http://www.nirim.go.jp/~izumi/rietan/angle_dispersive/angle_dispersive.html
http://www.ccp14.ac.uk/ccp/web-mirrors/rietan/~izumi/rietan/angle_dispersive/angle_dispersive.html
ftp://ftp.minerals.csiro.au/pub/xtallography/ccp14/ccp/web-mirrors/rietan/~izumi/rietan/angle_dispersive/angle_dispersive.html
Rietquan for Windows
Luca Lutterotti - Luca.Lutterotti@ing.unitn.it
Menu driven; concentrating on size/strain/stress analysis. Superceded by MAUD for Java.
http://www.ing.unitn.it/~luttero/
http://www.ccp14.ac.uk/ccp/web-mirrors/lutterotti/~luttero/
ftp://ftp.minerals.csiro.au/pub/xtallography/ccp14/ccp/web-mirrors/lutterotti/~luttero/index.html
Simref
Harold Ritter - harald.ritter@uni-tuebingen.de
Simultaneous Rietveld Refinement with Multible Powder Datasets High temperature experiment; Incommensurate structure refinement
Simpro Pawley software and MEED Maximum entropy also available.
http://www.uni-tuebingen.de/uni/pki/
http://www.ccp14.ac.uk/ccp/web-mirrors/pki/uni/pki/index.html
ftp://ftp.minerals.csiro.au/pub/xtallography/ccp14/ccp/web-mirrors/pki/uni/pki/index.html
WinMprof for Windows
Alain Jouanneaux - jouanneaux@univ-lemans.fr
Le Bail fitting. Has in built Fourier Q peak finding for building up structures.
http://pecdc.univ-lemans.fr/WinMProf/WinMProf.htm
http://www.ccp14.ac.uk/ccp/web-mirrors/winmprof/WinMProf/WinMProf.htm/
ftp://ftp.minerals.csiro.au/pub/xtallography/ccp14/ccp/web-mirrors/winmprof/WinMProf/WinMProf.htm
XND
Jean-Francois Berar - berar@polycnrs-gre.fr
Simultaneous Rietveld Refinement with Multible Powder Datasets High temperature experiment; Incommensurate structure refinement, Can handle "parasitic emission lines" such as CukB, Tungsten
http://RX-Crg1.polycnrs-gre.fr/public/xnd/xnd.html
ftp://labs.polycnrs-gre.fr/pub/xnd/
http://www.ccp14.ac.uk/ccp/web-mirrors/xnd/public/xnd/xnd.html
http://www.ccp14.ac.uk/ccp/ccp14/ftp-mirror/xnd/pub/xnd/
ftp://ftp.minerals.csiro.au/pub/xtallography/ccp14/ccp/web-mirrors/xnd/public/xnd/xnd.html
ftp://ftp.minerals.csiro.au/pub/xtallography/ccp14/ccp/ccp14/ftp-mirror/xnd/pub/xnd/
XRS-82/DLS-76
Christian Baerlocher - baerlocher@kristall.erdw.ethz.ch
Structure Refinement, ASCII Control Files
http://kristall.erdw.ethz.ch/software/soft.html
http://www.ccp14.ac.uk/ccp/web-mirrors/ethz/software/soft.html
ftp://ftp.minerals.csiro.au/pub/xtallography/ccp14/ccp/web-mirrors/ethz/software/soft.html
Rietveld Program Interfaces
Interfaces into Rietveld programs vary from GUIs to direct editing of ASCII files.
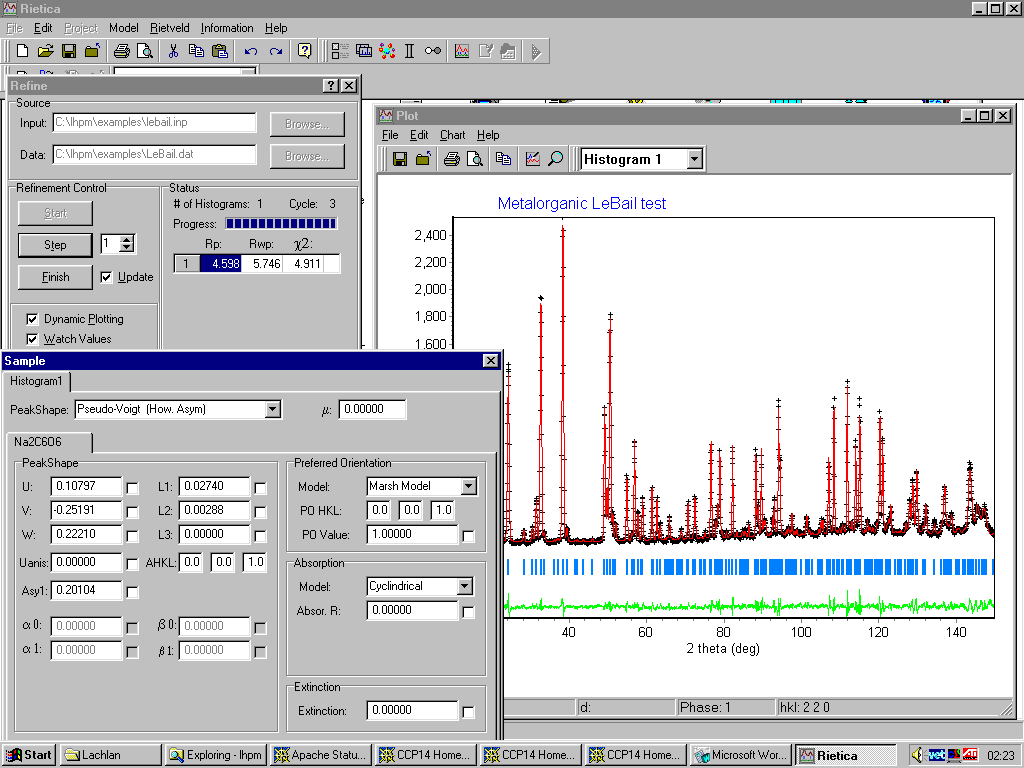
GUI LHPM/Rietica for Windows
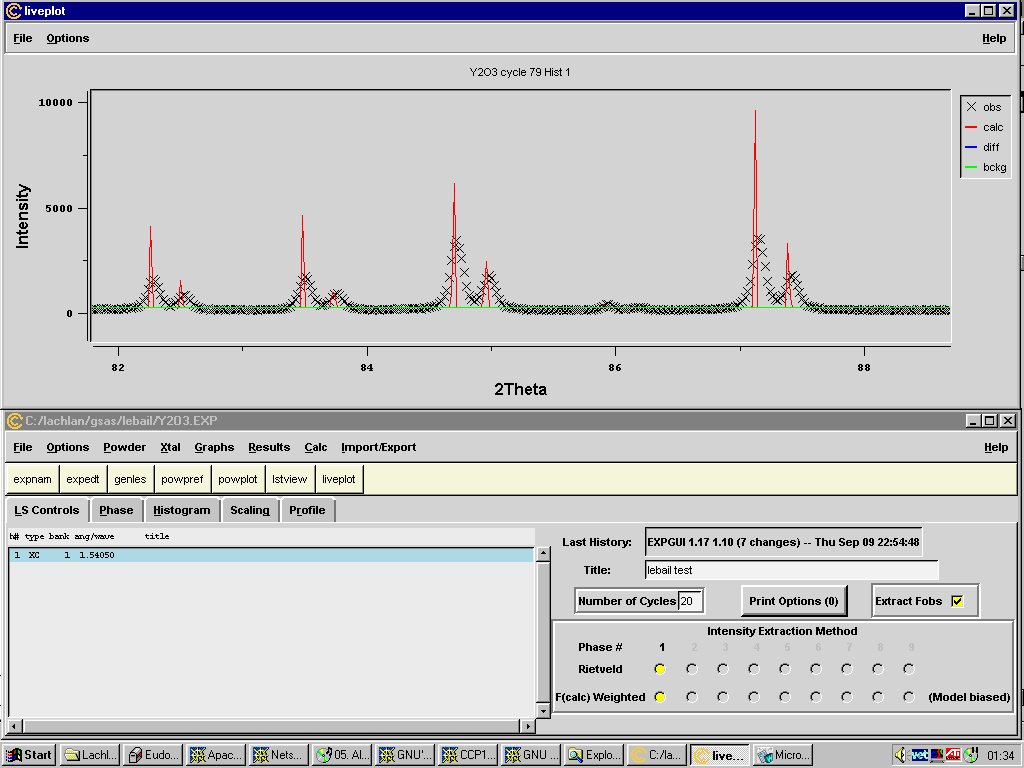
EXPGUI for GSAS:
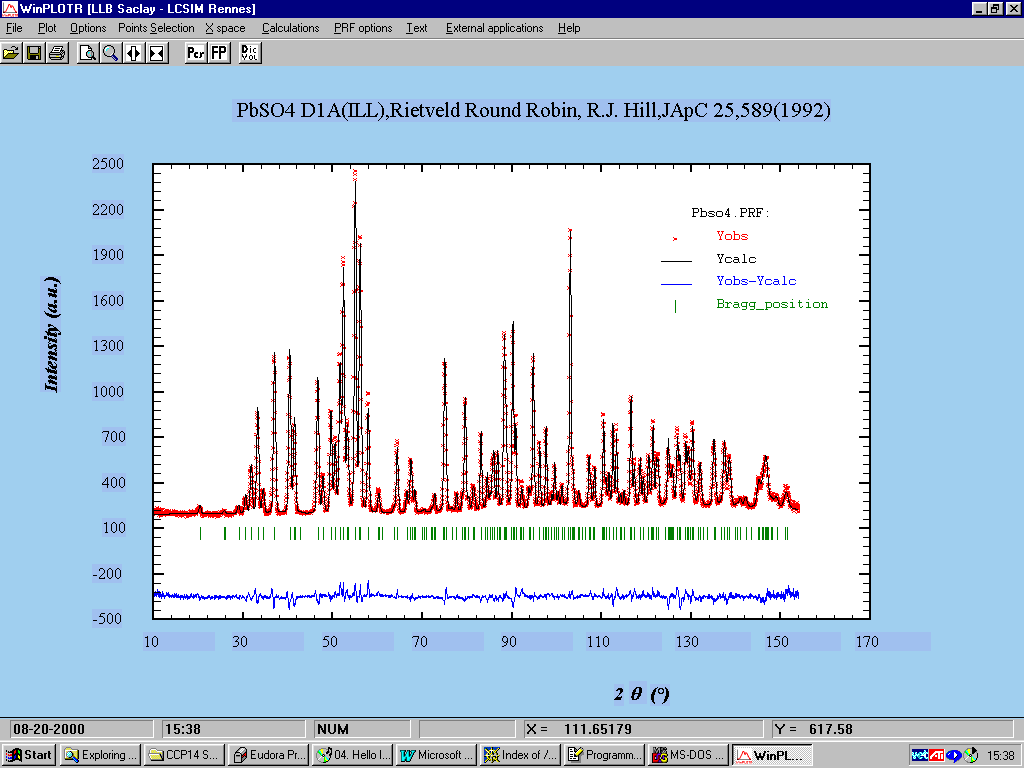
Fullprof
Where Rietveld programs are "behind" compared to Single Crystal Refinement Suites.
(focussing on organic structures)
Restrained Rietveld Structure Refinement
(Organics)
Software not as powerful as single crystal but there are some tutorials with tricks on the CCP14 web site:
http://www.ccp14.ac.uk/solution/restrained_rietveld/
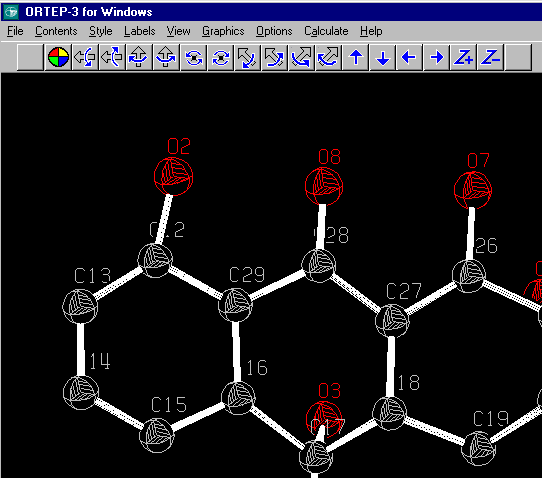
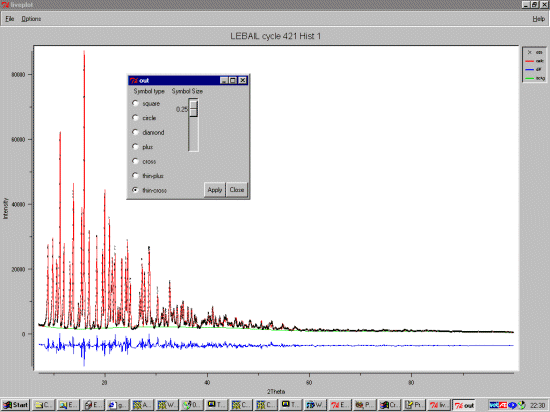
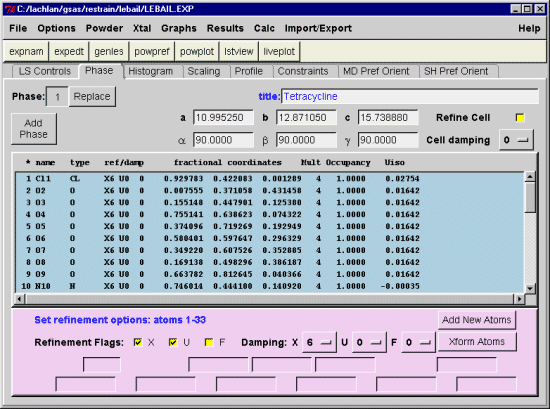
Single Crystal Suites
(Applicable to Powder Diffraction: validation, etc )
Single crystal suites can be very powerful
as they allow users to quickly pick and mix a variety of programs at the click of a button.Range of programs to choose from:
http://www.ccp14.ac.uk/solution/xtalsuites/
Crystals –Watkin, Prout, Carruthers, Betteridge, R.I. Cooper
Shelxs and Sir for Structure Solution
Crystals for refinement
Fourier Map Calculation and Viewing
Guided refinement for students and chemists.
http://www.xtl.ox.ac.uk/
CCP14 Mirror: http://www.ccp14.ac.uk/ccp/web-mirrors/crystals/
DS*SYSTEM
ShakePSD, ShakePSDL, Multan for Structure Solution
LSBF for refinement
http://www.ccp14.ac.uk/ccp/web-mirrors/okada/
ORTEX - Patrick McArdle
Shelx86/Shelx97 for Structure Solution
Shelxl for refinement
http://www.nuigalway.ie/cryst/software.htm
CCP14 Mirror: http://www.ccp14.ac.uk/ccp/web-mirrors/ortex/cryst/software.htm
Single Crystal Suites (Cont’d)
(Applicable to Powder Diffraction)
System S/Platon - Ton Spek
Shelx86/Shelx97, Sir97, Dirdif, Crunch Structure Solution
(Crunch, EXOR, Sir97 and Dirdif for auto-building)
Shelxl for refinement
Fourier Map Calculation and Viewing
Fully automatic Structure solution and refinement
http://www.cryst.chem.uu.nl/platon/
CCP14 Mirror: http://www.ccp14.ac.uk/ccp/web-mirrors/platon-spek/software.html
WinGX - Louis Farrugia
Shelx86/Shelx97, Sir92/Sir97, Dirdif, Patsee for Structure Solution
Shelxl for refinement
Fourier Map Calculation and Viewing
Links to very large array of programs.
http://www.chem.gla.ac.uk/~louis/wingx/
CCP14 Mirror: http://www.ccp14.ac.uk/ccp/web-mirrors/farrugia/~louis/wingx/
Xtal (GPL’d)
Crisp and Patsee for Structure Solution
Xtal for refinement
Fourier Map Calculation and Viewing
http://xtal.crystal.uwa.edu.au/
CCP14 Mirror: http://www.ccp14.ac.uk/ccp/web-mirrors/xtal/
NRCVAX
Solver for Structure Solution
NCVAX for refinement
Contact: pwhite@pyrite.chem.unc.edu
Single Crystal Suite Examples
WinGX menu system:
E.g., Links to Shelxs 86/97, Sir 92/97, Dirdif, Patsee and major number of other programs and utilities including Platon.
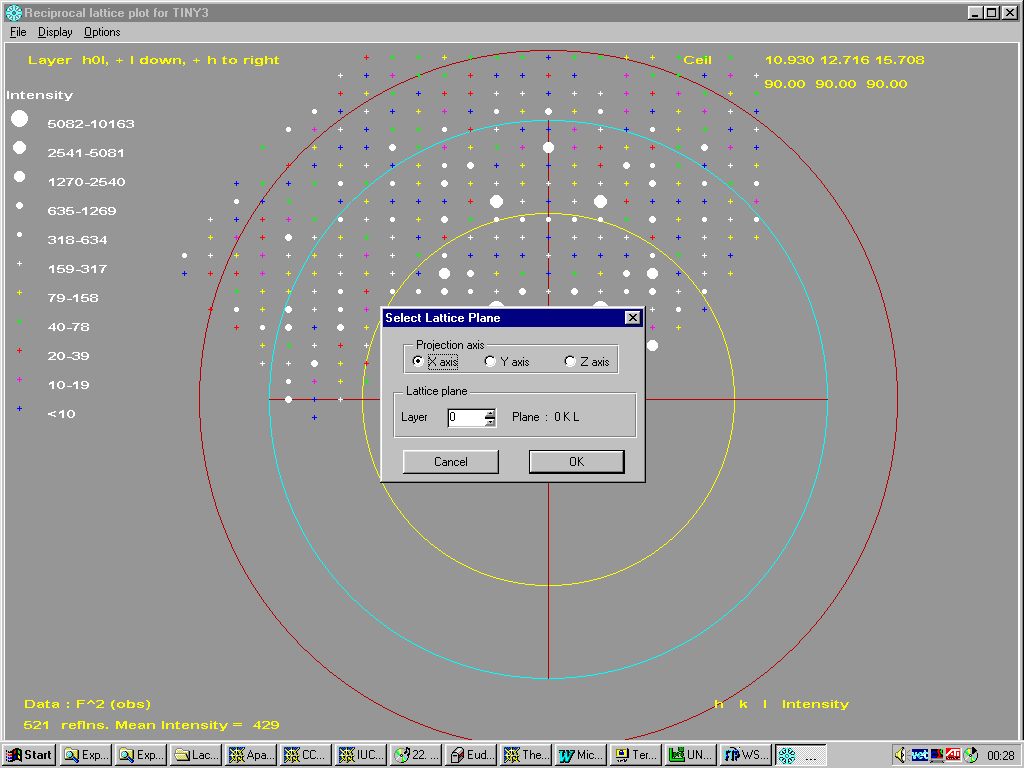
Platon/System S menu system for UNIX:
E.g., Links to Shelxs 86/97, Sir97, Crunch and Dirdif and multiple internal applications
Multiple Pathways for Structure Solving
Solving in one program – automatic building in another: via Platon/System S
(Cesium Titanium Silicate)
Solve in Shelx86
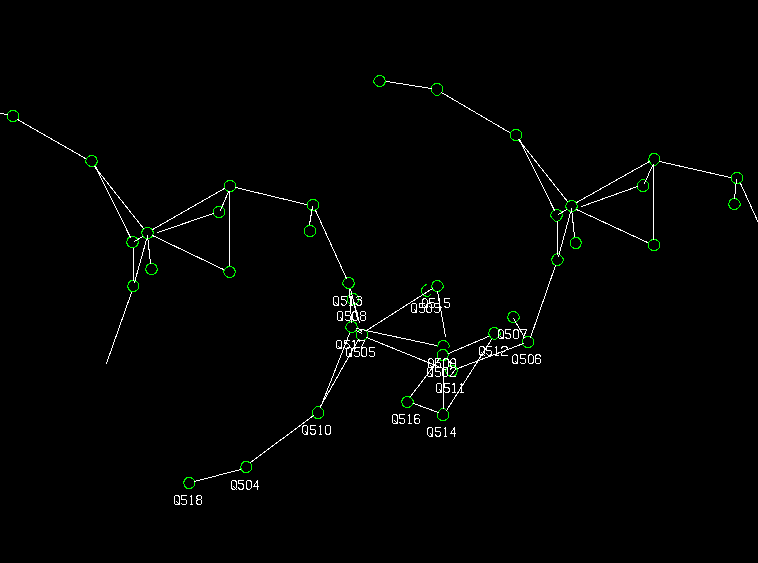
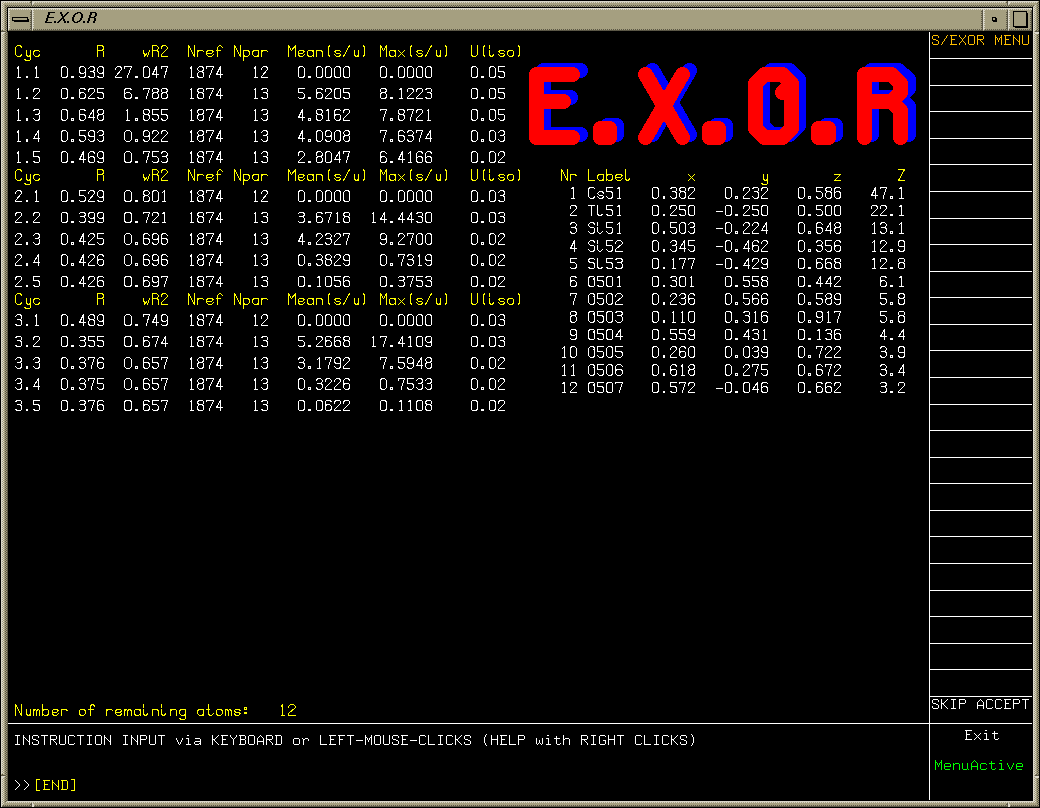
Use the System S/Platon EXOR function to build up the structure
(also have the other options of using Crunch, Sir and Dirdif to build up the structure).
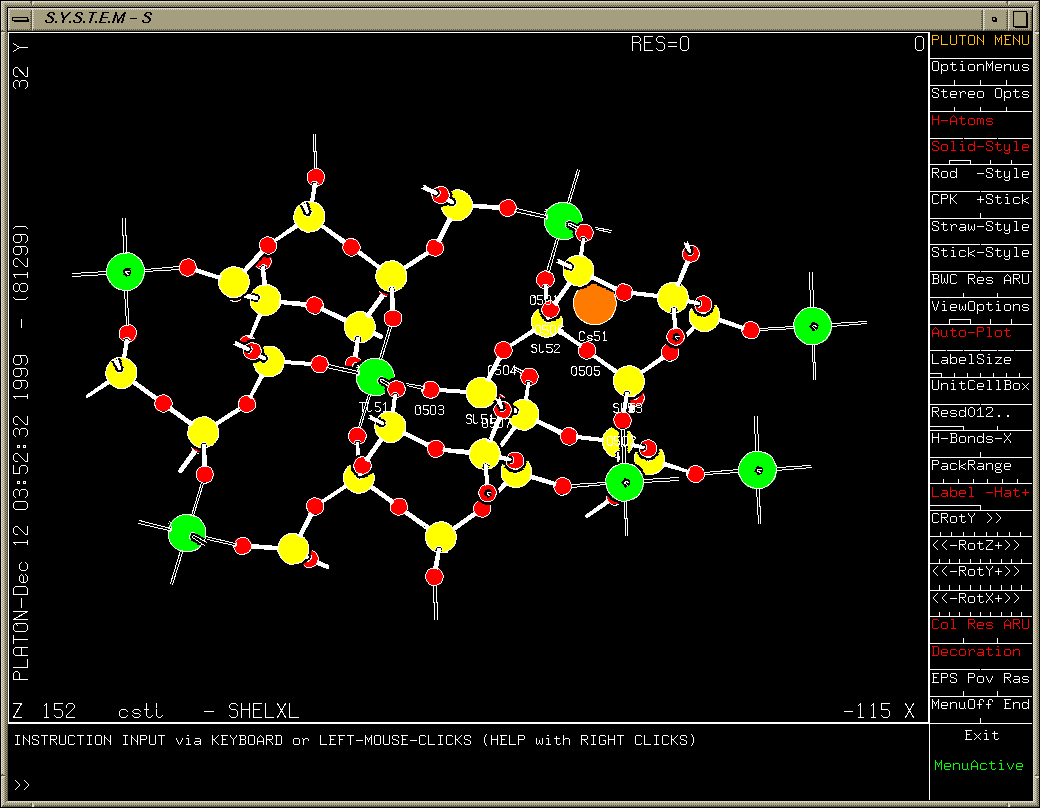
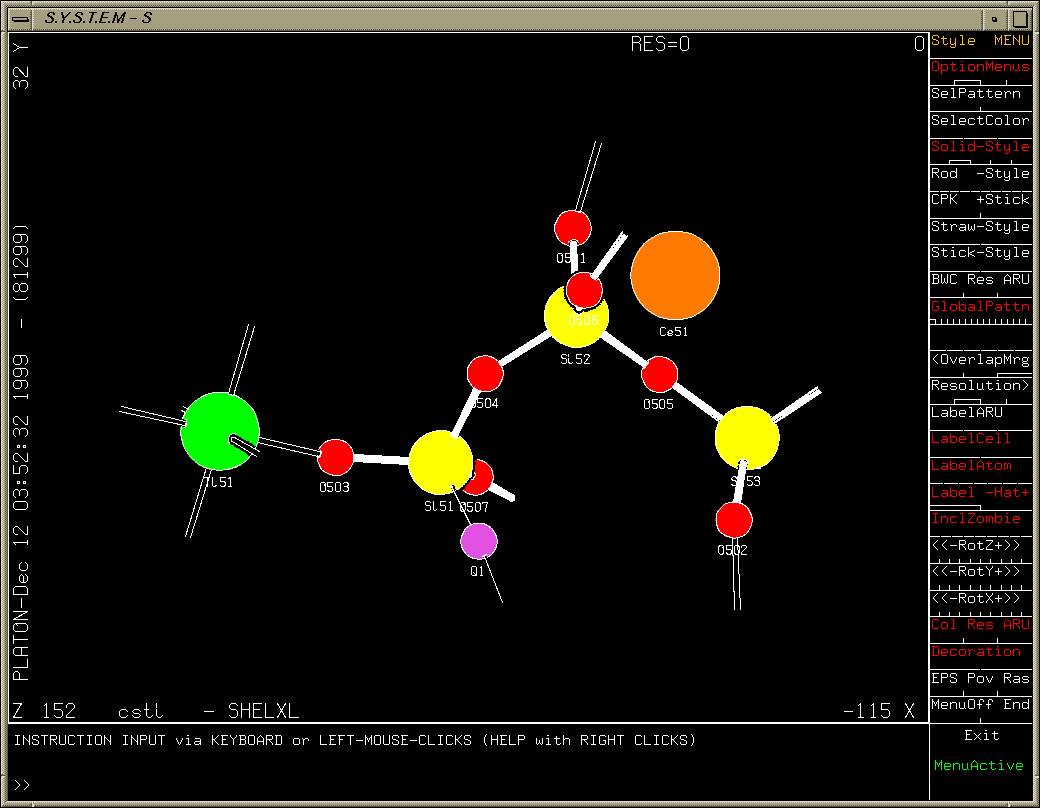
One oxygen atom left to find (after refining heavy atoms anisotropically). Purple Q peak stands out quite clearly.
Transparently Running the Squeeze Option (that comes with Platon) within WinGX
SQUEEZE: An effective cure for the disordered solvent syndrome in crystal structure refinement.
http://www.cryst.chem.uu.nl/platon/pl000303.html
Graphically Interacting with the Structure
Most freely available programs and suites now have the ability to interact directly with a graphical representation of the structure during solving and refining.
For routine and not so routine refinements, point and click interfaces are now available, complementing the ASCII file or command line interfaces.
3rd party programs can be useful: e.g
., Ortep-3 by Louis Farrugia will also read Fullprof, Rietica and GSAS Rietveld structure files.Graphically Interacting with the Structure
Crystals (http://www.xtl.ox.ac.uk/)
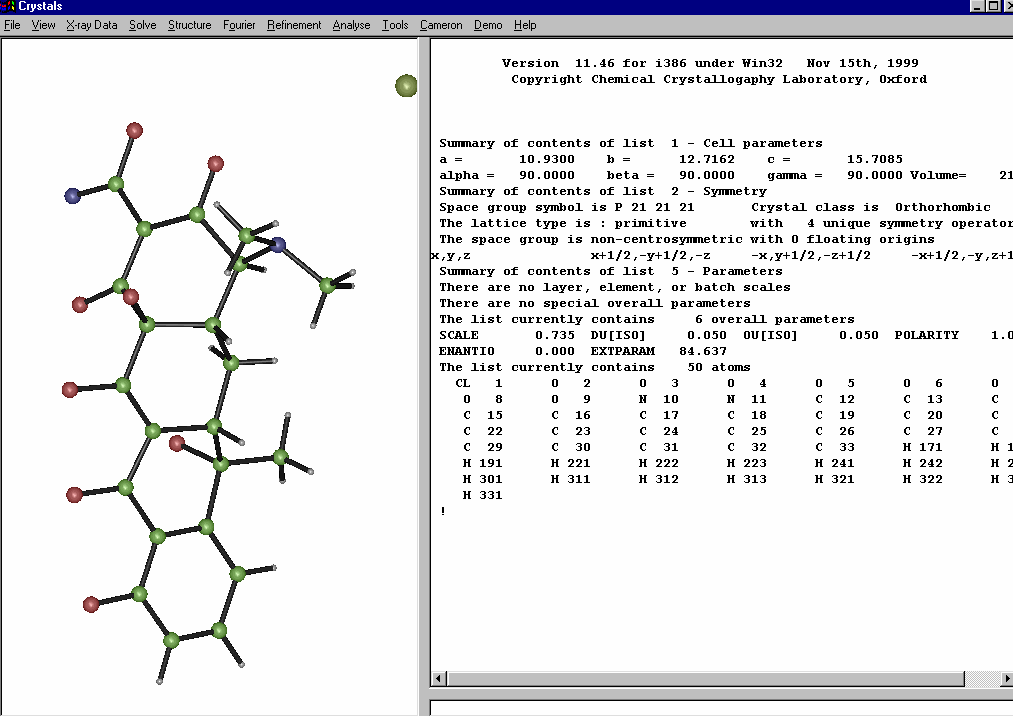
Cameron (comes with Crystals and WinGX)
http://www.xtl.ox.ac.uk/
http://www.chem.gla.ac.uk/~louis/wingx/
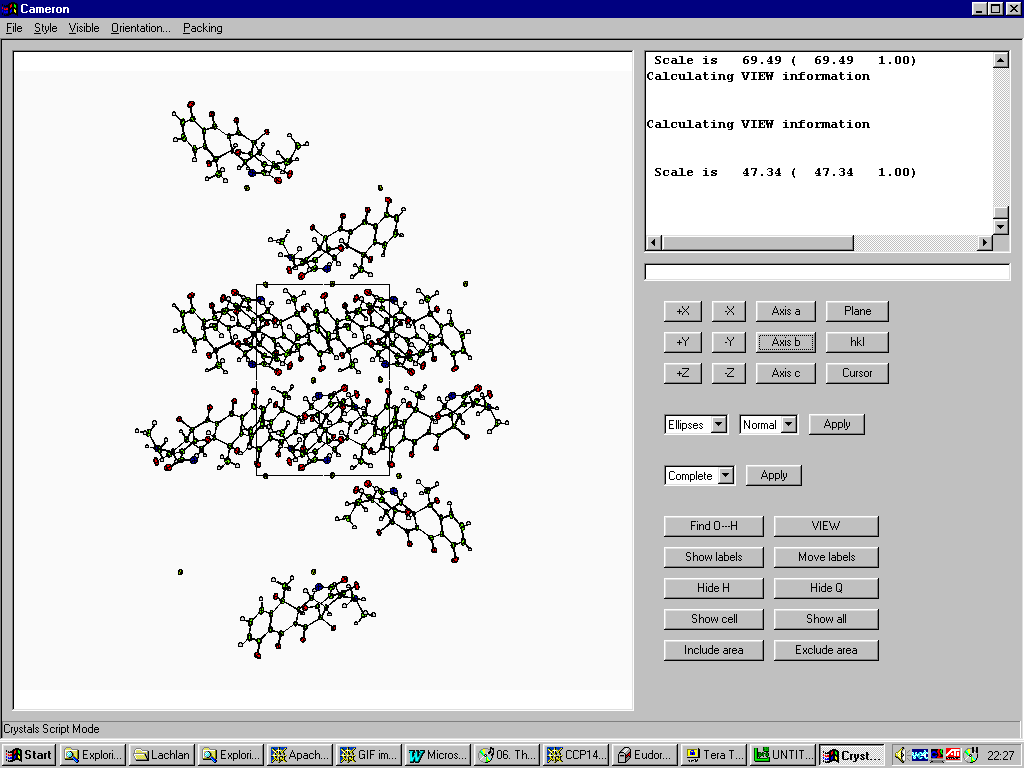
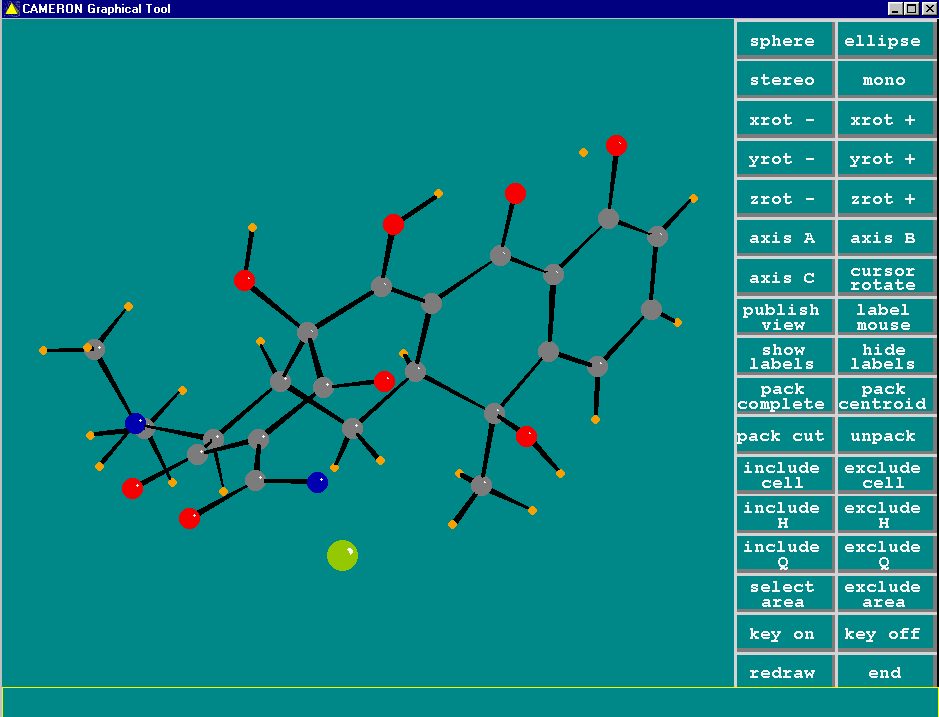
Graphically Interacting with the Structure
PIG (part of the Xtal suite)
http://xtal.crystal.uwa.edu.au/
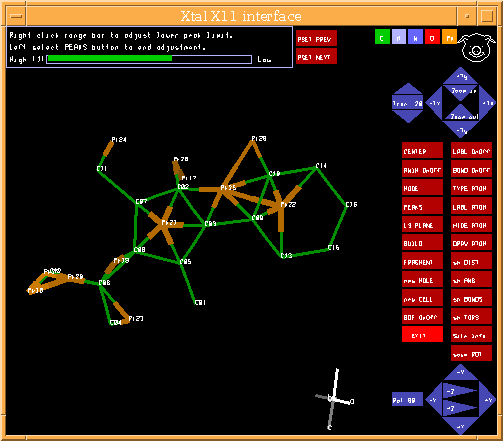
ORTEX
http://www.nuigalway.ie/cryst/software.htm
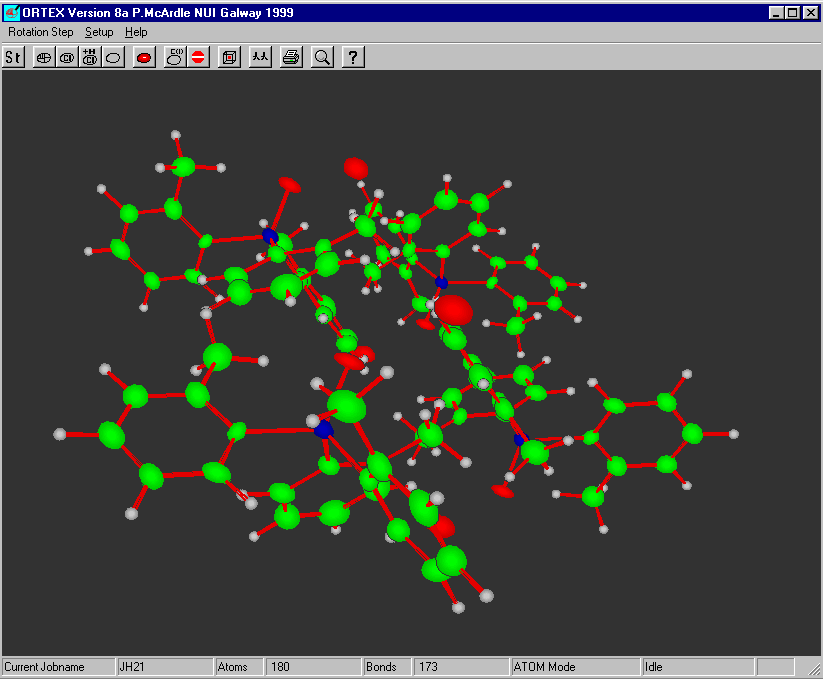
Graphically Interacting with the Structure
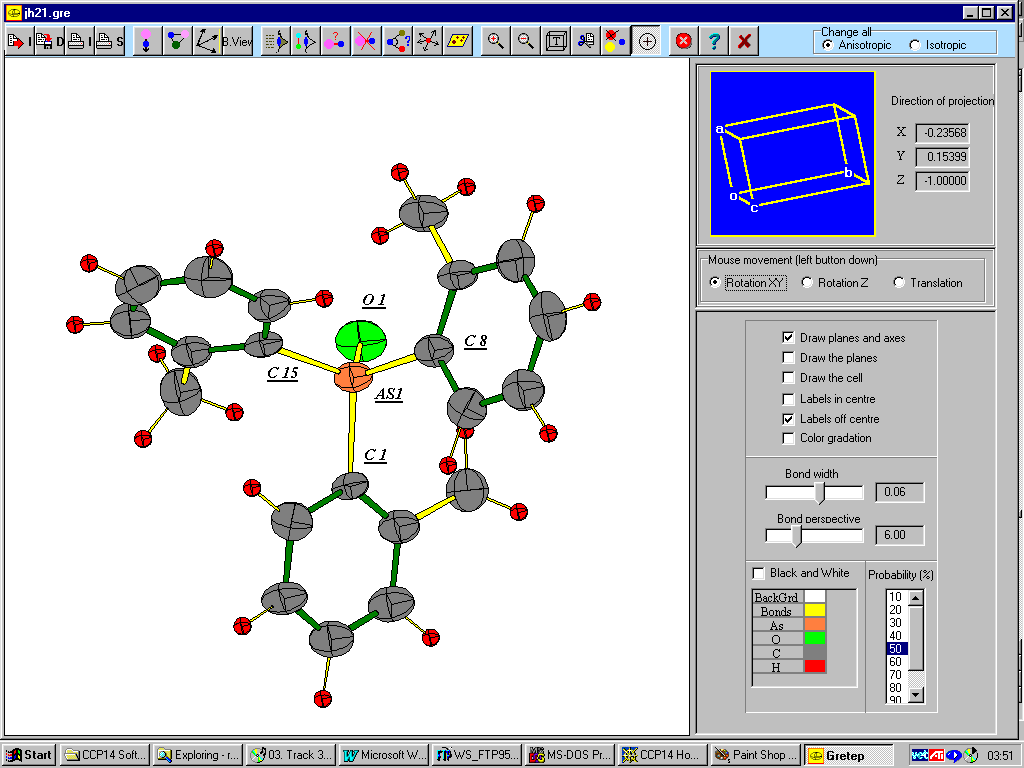
Gretep
http://www.ccp14.ac.uk/tutorial/lmgp/#gretep
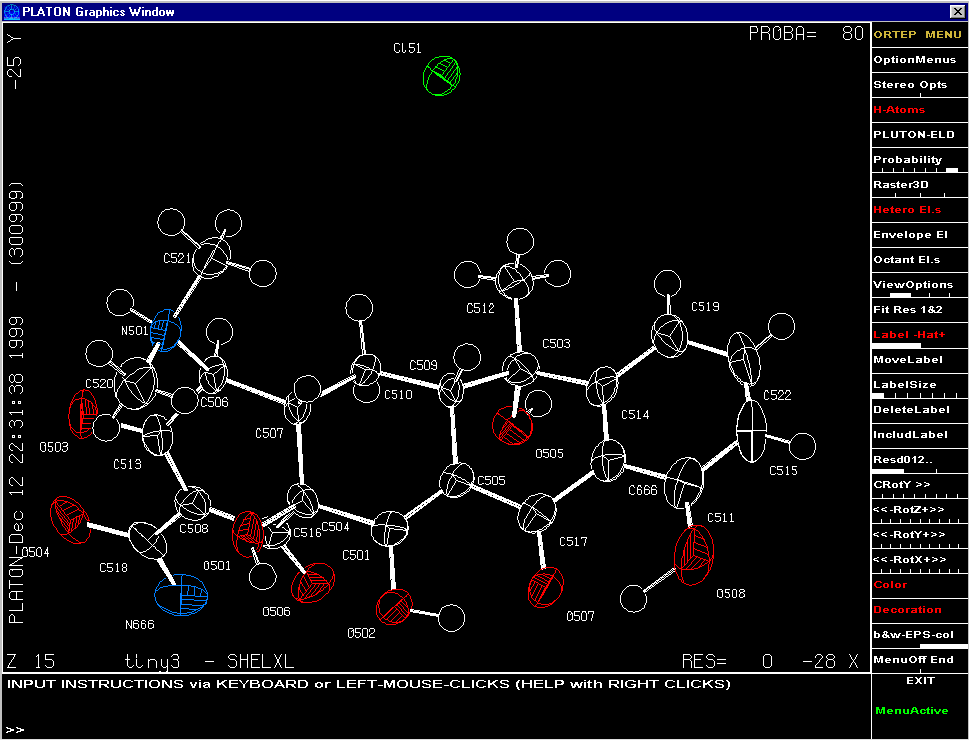
Platon (in ADP ORTEP Mode)
UNIX: http://www.cryst.chem.uu.nl/platon/
Windows: http://www.chem.gla.ac.uk/~louis/platon97/
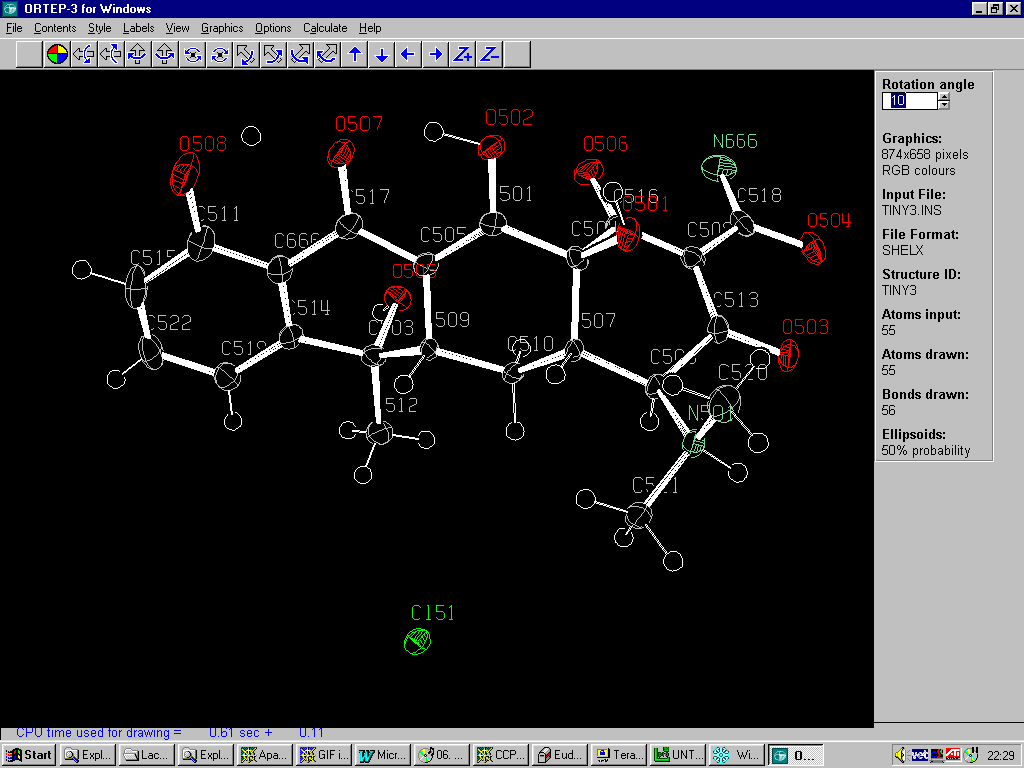
Graphically Interacting with the Structure
Ortep-3
http://www.chem.gla.ac.uk/~louis/ortep3/
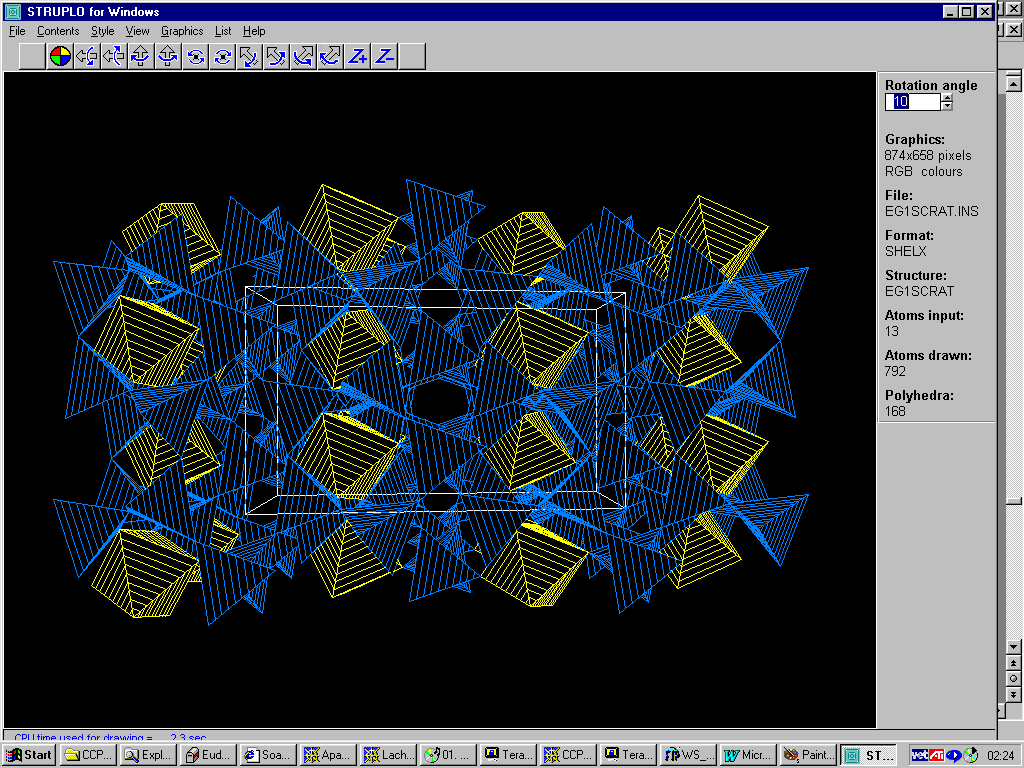
GUI WinSTRUPLO
http://www.chem.gla.ac.uk/~louis/struplo/
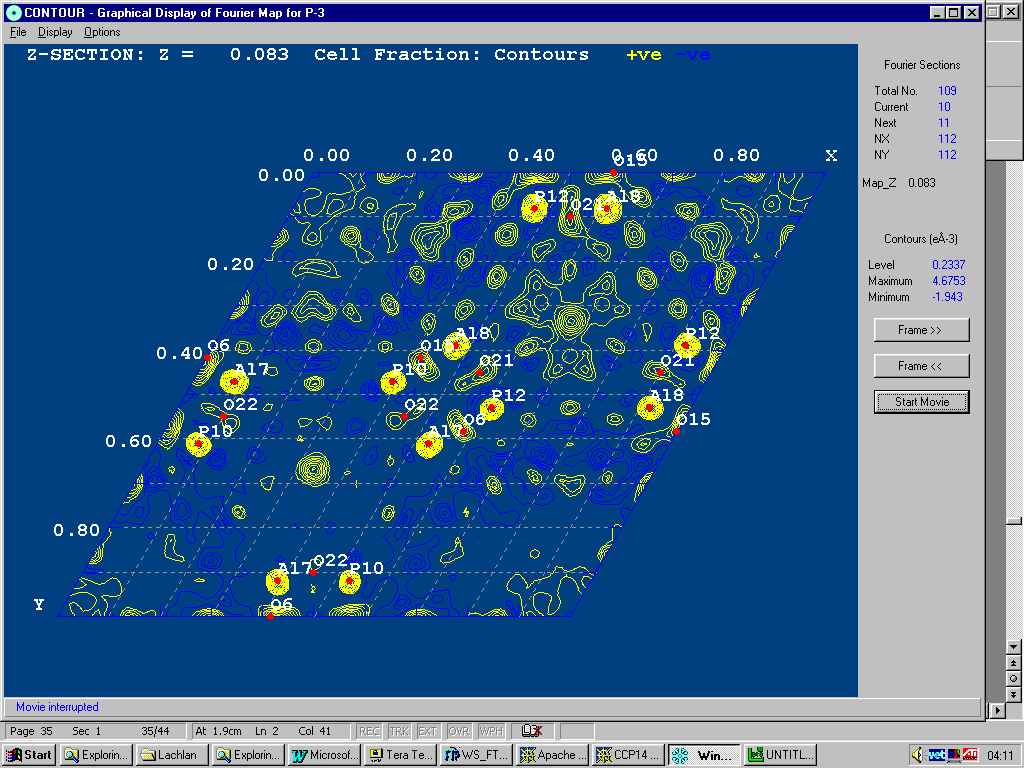
Fourier Capability in Rietveld Software (
ability to effectively refine structures is falling behind abilty to solve– not making use of optimal algorithms such as Maximum Entropy )Summary List at:
http://www.ccp14.ac.uk/solution/rietveld_fourier_maps/
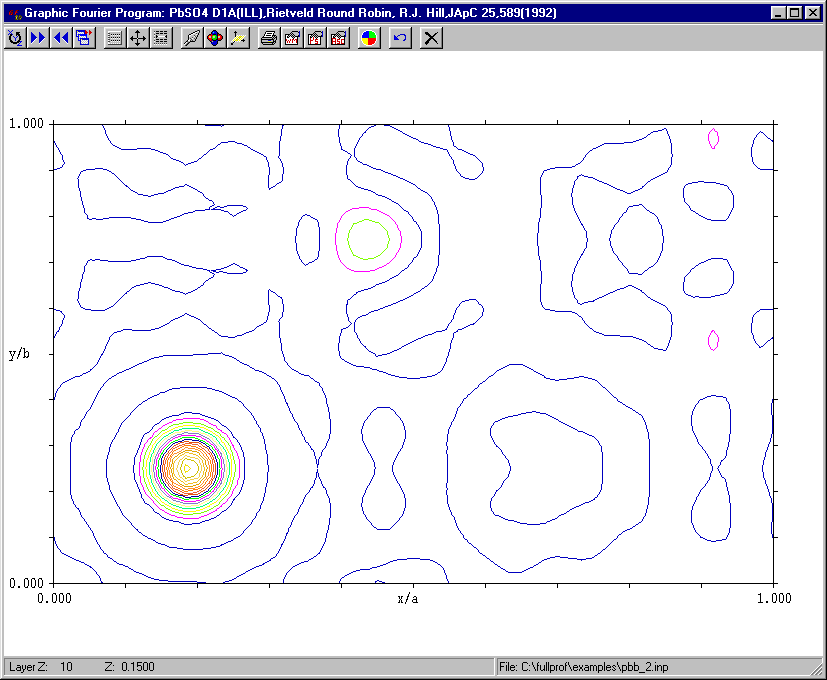
Examples:
Fullprof/GFOUR
GSAS
(including
VRML output)
WinMPROF (Q peak location)
Single Crystal Suite Fourier Map Generators and Viewers
(applicable for Powder Diffraction)
Foue (GPL’d) by Scott Belmonte can convert GSAS Fourier Maps into WinGX/Crystals and Project XD format)
http://www.ccp14.ac.uk/ccp/web-mirrors/scott-belmonte-software/
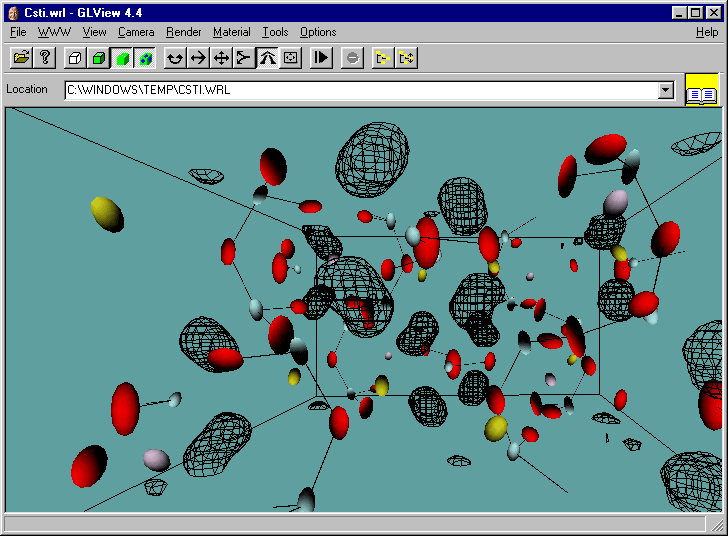
Example of WinGX fourier map animating through a zeolite with disordered template.
Fourier Maps – Single Crystal Suites
Crystals – Slant Plane Fourier Map:
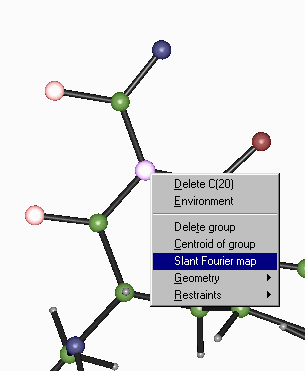 Define the Slant Plane by selecting 3 atoms then right click to go into the Fourier Map option.
Define the Slant Plane by selecting 3 atoms then right click to go into the Fourier Map option.
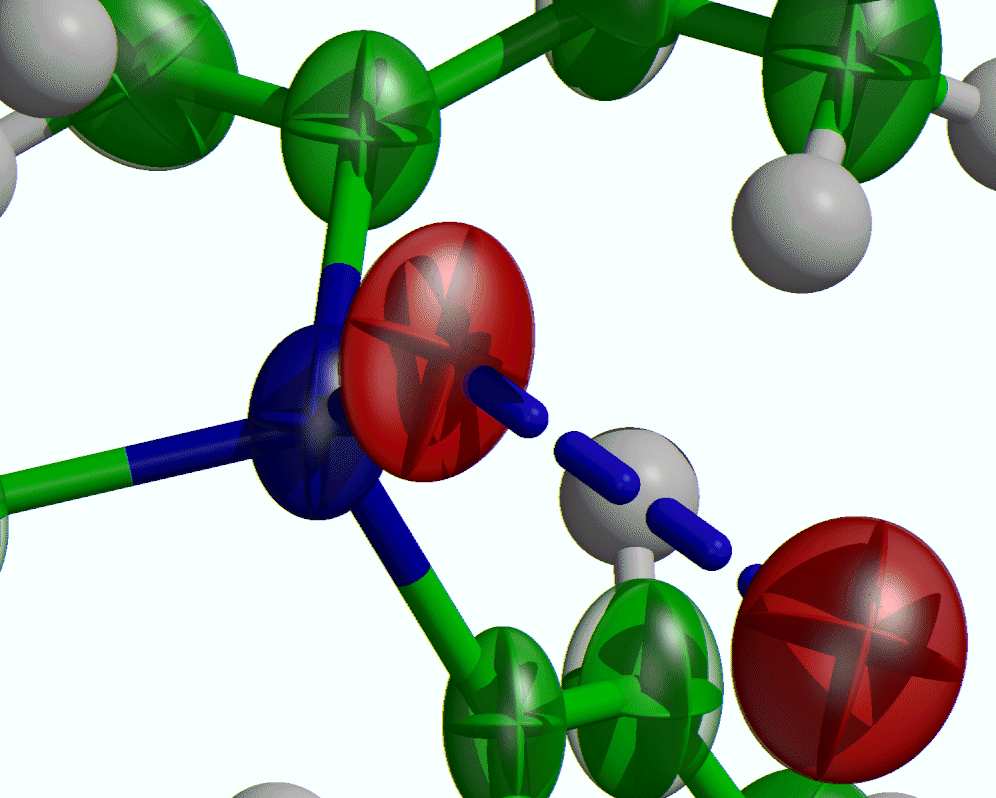
Examine slant plane difference map looking at hydrogen positions – via the Marching Cubes 3D Fourier Map viewer by Michal Husak (also linked into)
http://mysak.umbr.cas.cz/~husakm/Public/MarchingCubeELD/MarchingCubeELD.htm
http://www.ccp14.ac.uk/ccp/web-mirrors/marchingcube-fourierviewer/~husakm/Public/MarchingCubeELD/MarchingCubeELD.htm
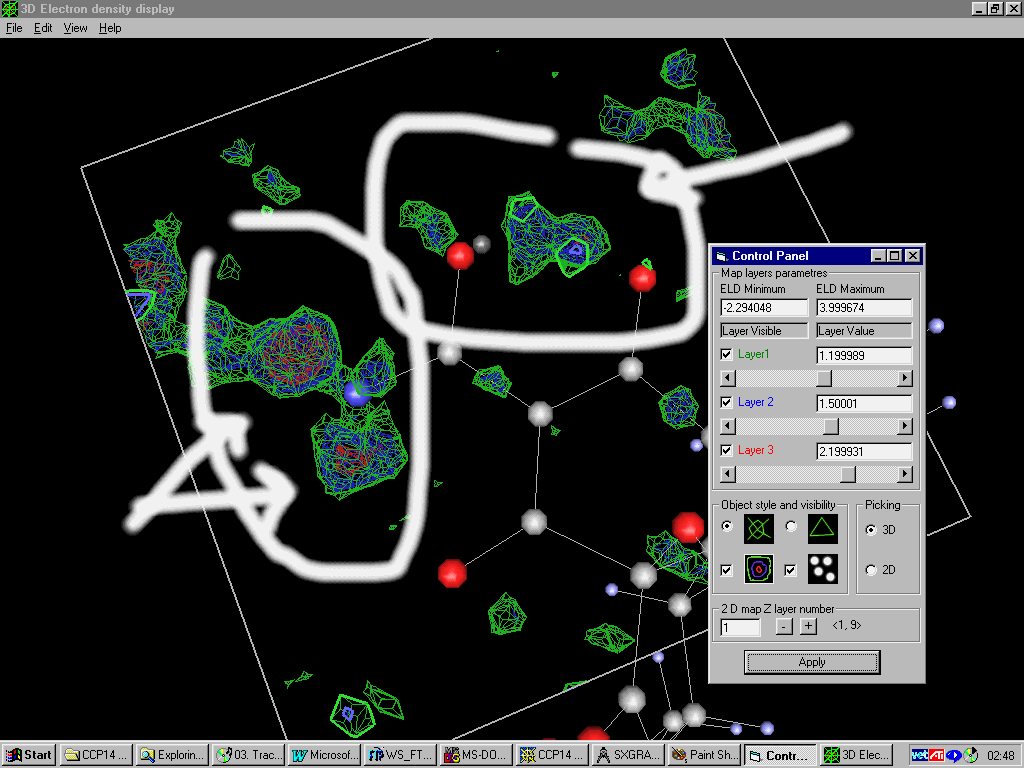
Platon/System S Fourier Map:
Examining slant plane difference map looking at hydrogen positions.
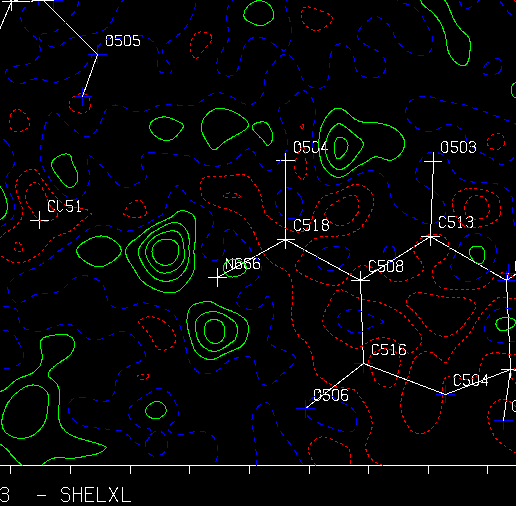
Using WinGX MapView:
(Can be run as a freestanding program linked into any program that will output a Mapview friendly format)
Examining slant plane difference map looking at hydrogen positions
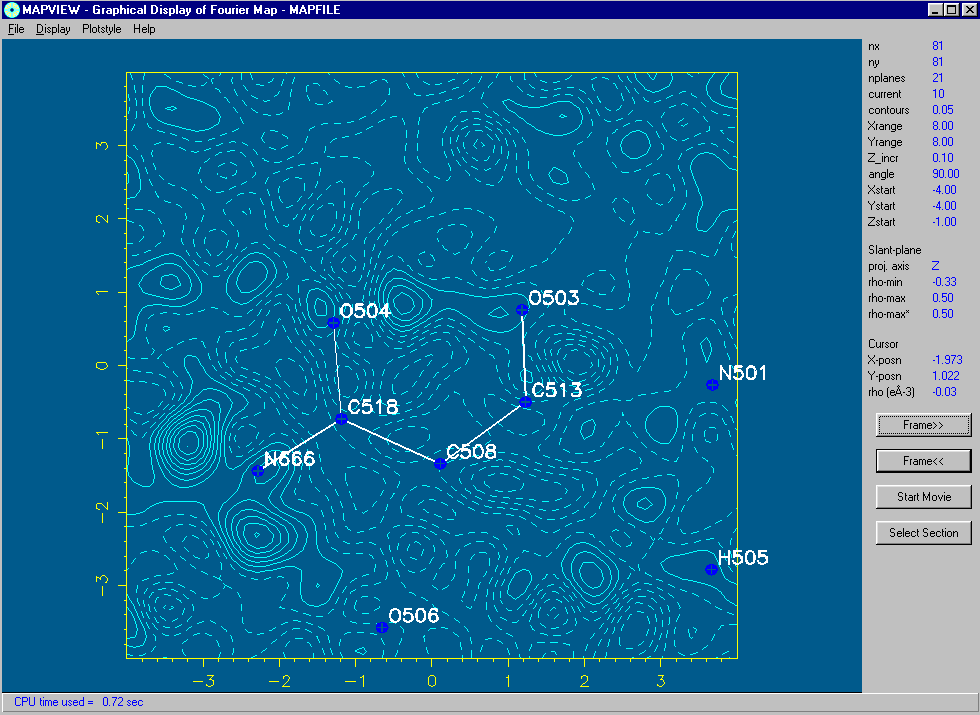
Using the 2D Bitmap Mode of MapView:
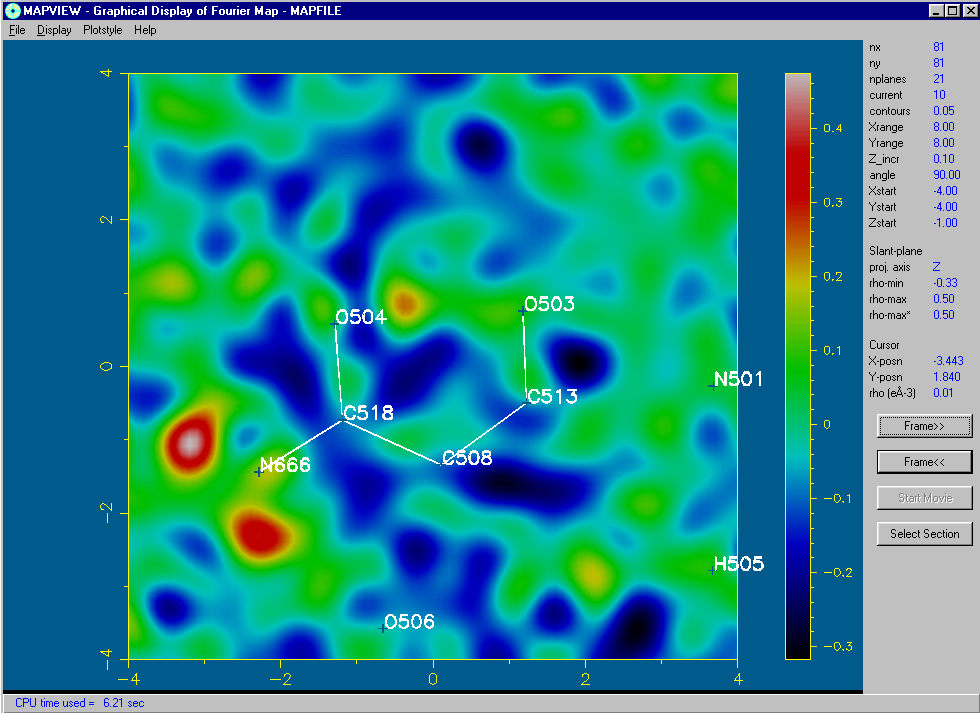
Structure Quality Checking
(Applicable to Powder Diffraction)
(Each suite can offer different features)
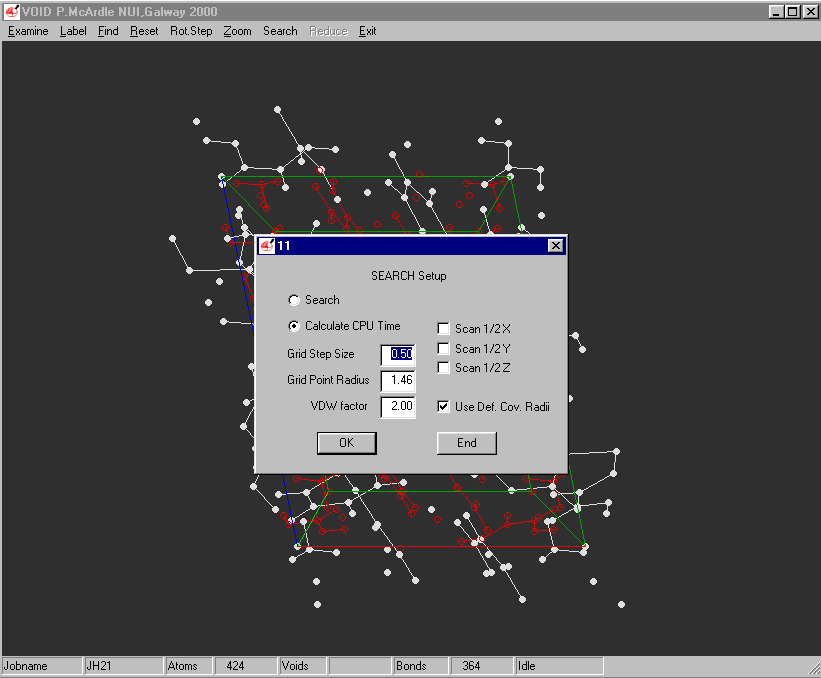
ORTEX:
Example of the Void Finding and graphical viewing within ORTEX (including estimate of time to completion)
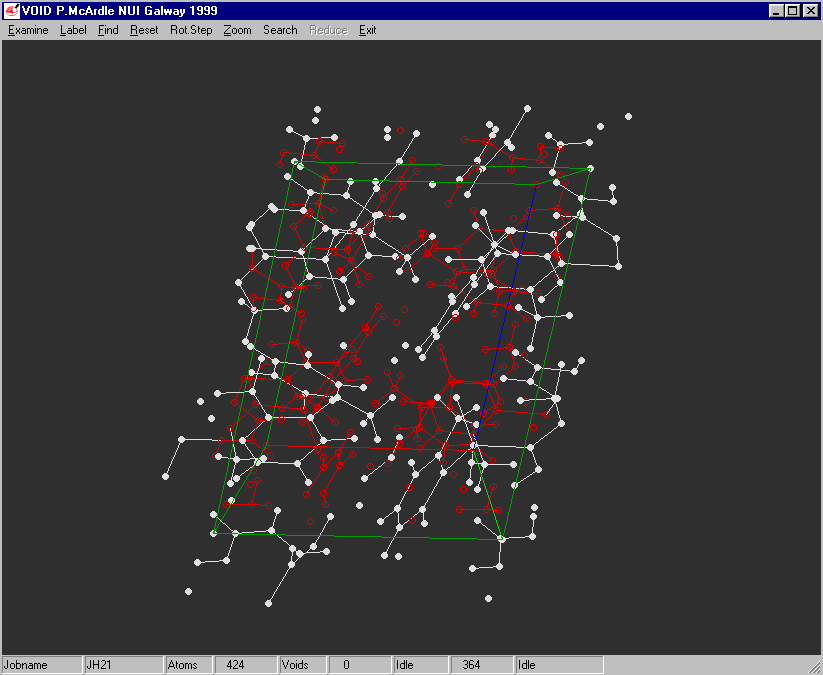
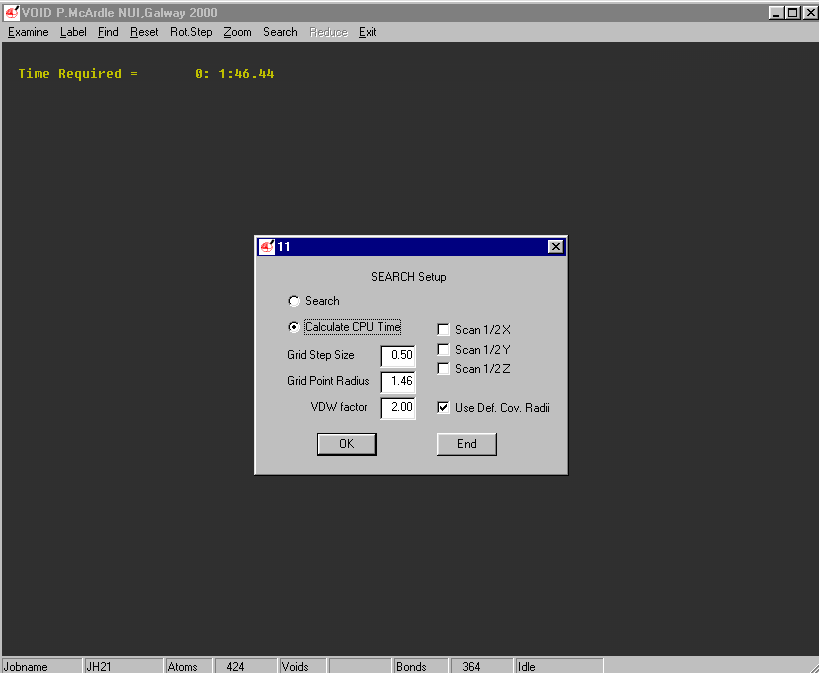
Structure Quality Checking
WinGX:
Has GUIs linking to a wide variety of Structure Checking, Analysis Options and Utilities (including a Windows version of Platon).
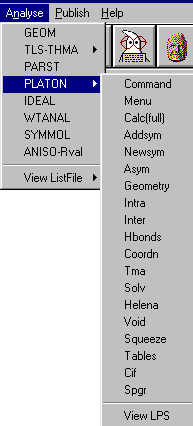
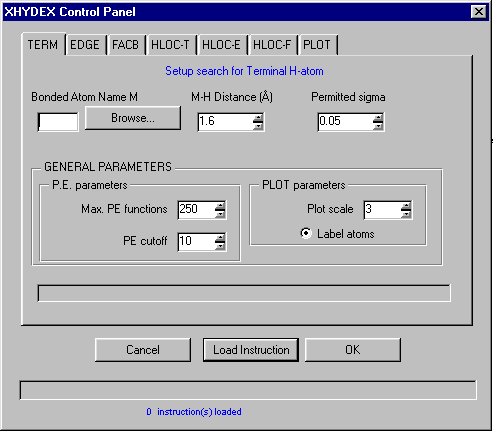
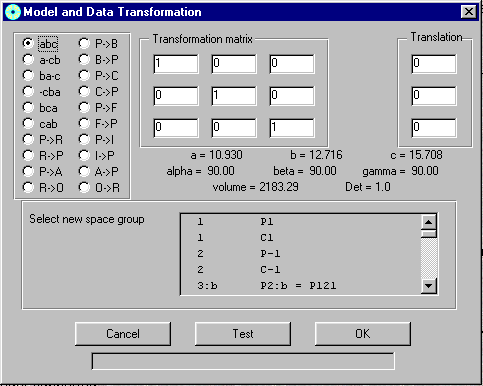
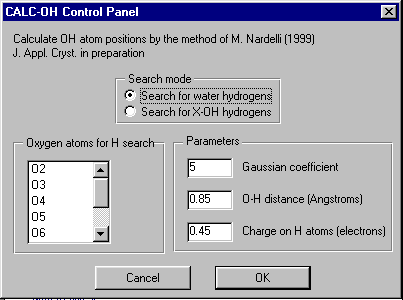
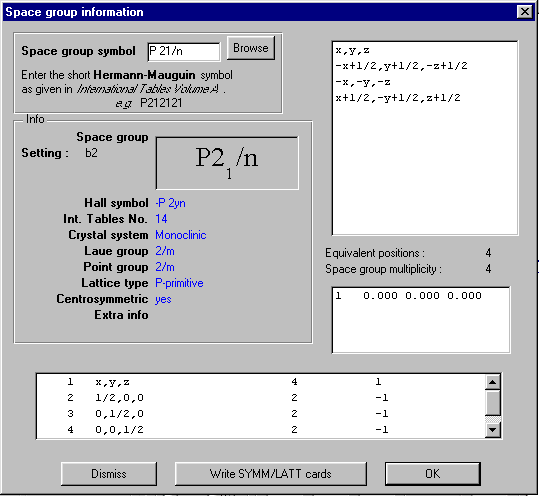
GUI XHYDEX (GUI over code by G. Orpen): Searching for Likely hydrogen positions. GUI CALC-OH (Nardelli 1999) GUI Parst, GUI Model and Data Transformation, SG Info, etc, etc.
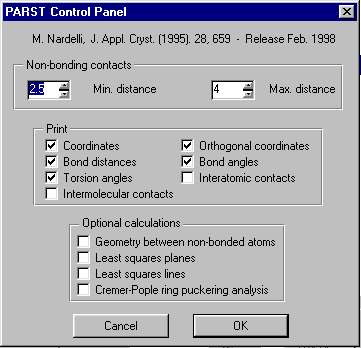
Platon (including Addsym - a mandatory program for all crystallographers!):
(links into WinGX and Crystals suite for Windows)
Addsym: check for missing symmetry. Will display or output a modified Shelx file with the updated structure if it finds a higher symmetry spacegroup.
Will report the miss-fitting atoms!
Refer: Short Communication: "P1 or P-1? Corrigendum", Acta Cryst B56 (2000) 744 from Richard E. Marsh
Original Structure Published in 1997 Triclinic, P1
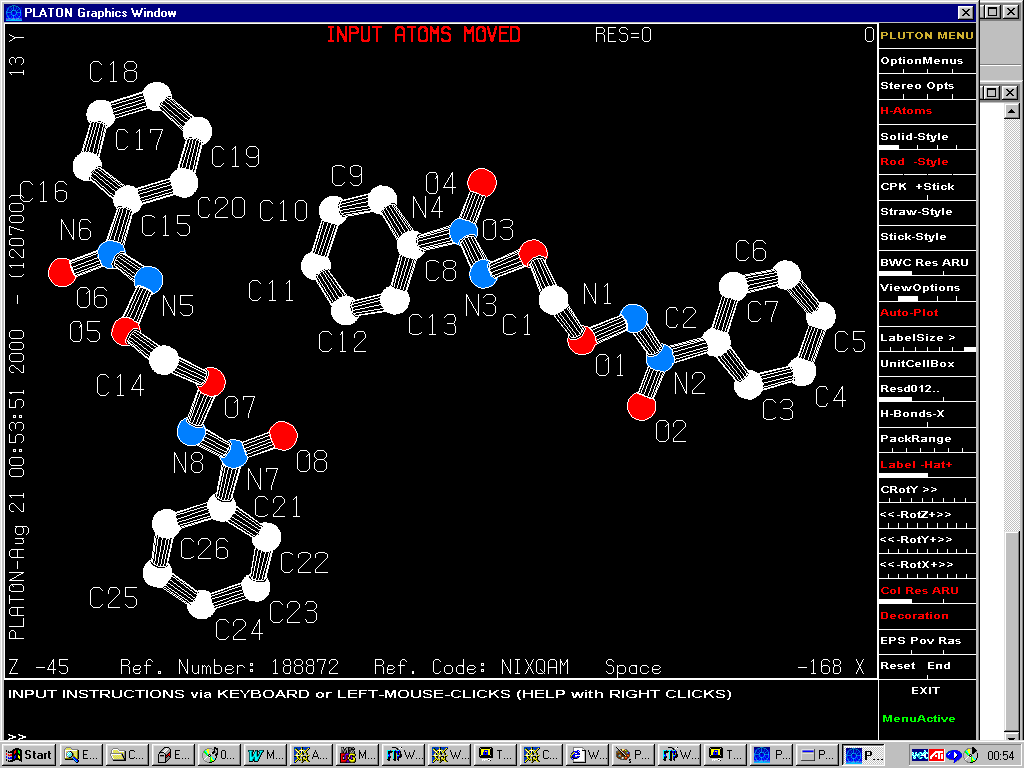
Reinterpretation by Marsh in 1999
Gives: Monoclinic C2
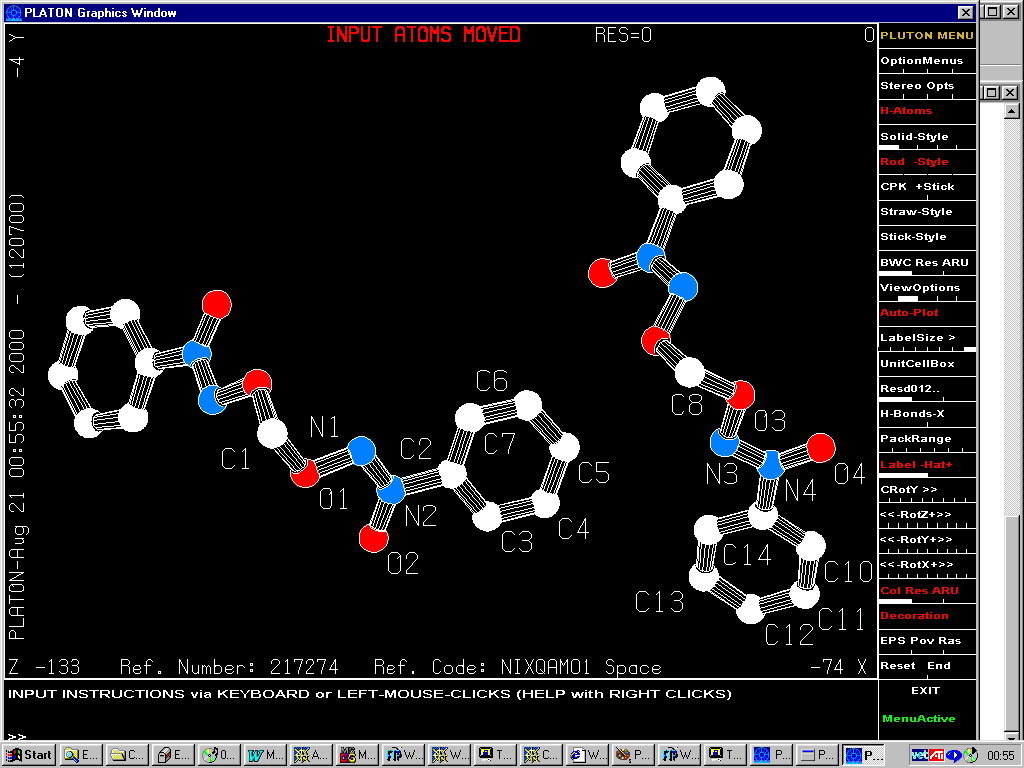
Press of a button in Platon/Addsym
(on either structure) gives:
Orthorhombic Fdd2
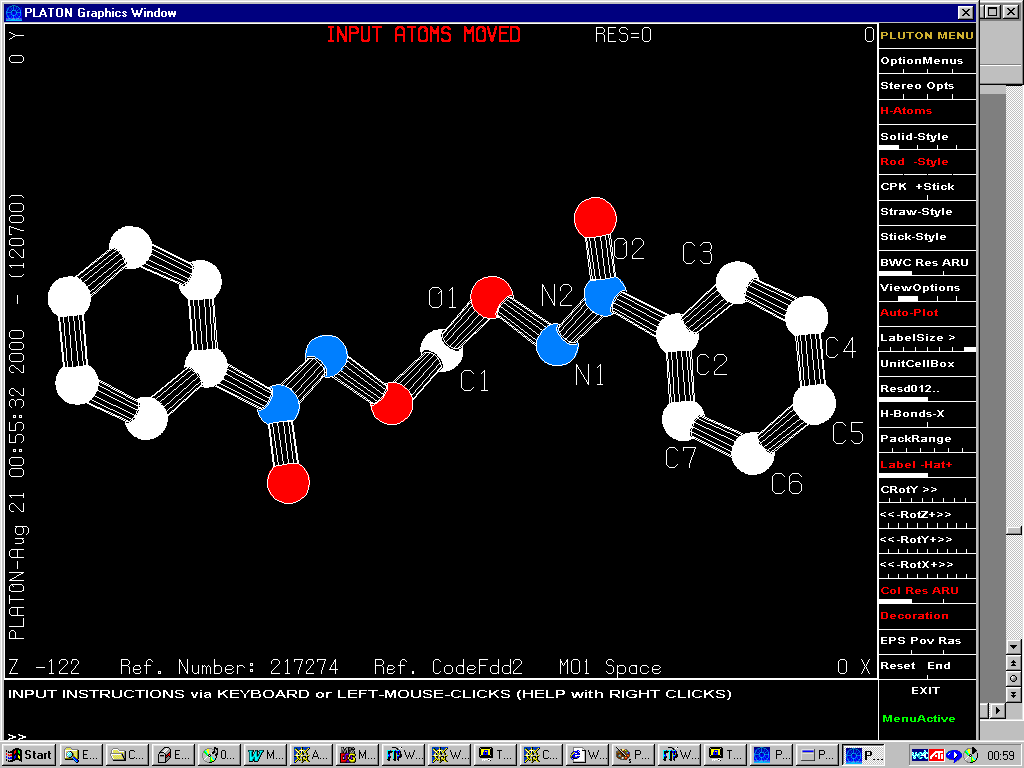
(with the option of generating a new Shelx *.RES file)
Is trivial to run Addsym routinely via Platon
Another Example of Platon/Addsym
(> Nov 2000 version)
http://www.cryst.chem.uu.nl/platon/pl000401.html
PLATON/ADDSYM analysis of all 1970 Cc structures in the CSD (Apr 1999):
http://www.cryst.chem.uu.nl/platon/cc.mis
PLATON/ADDSYM analysis of all 1848 P1 structures in the CSD (Oct 1999):
http://www.cryst.chem.uu.nl/platon/p1.mis
A Case of subcell symmetry
Originally in P1:
(96 independent Carbon atoms)
9.74832 12.74268 15.70341 99.506 77.047 92.504
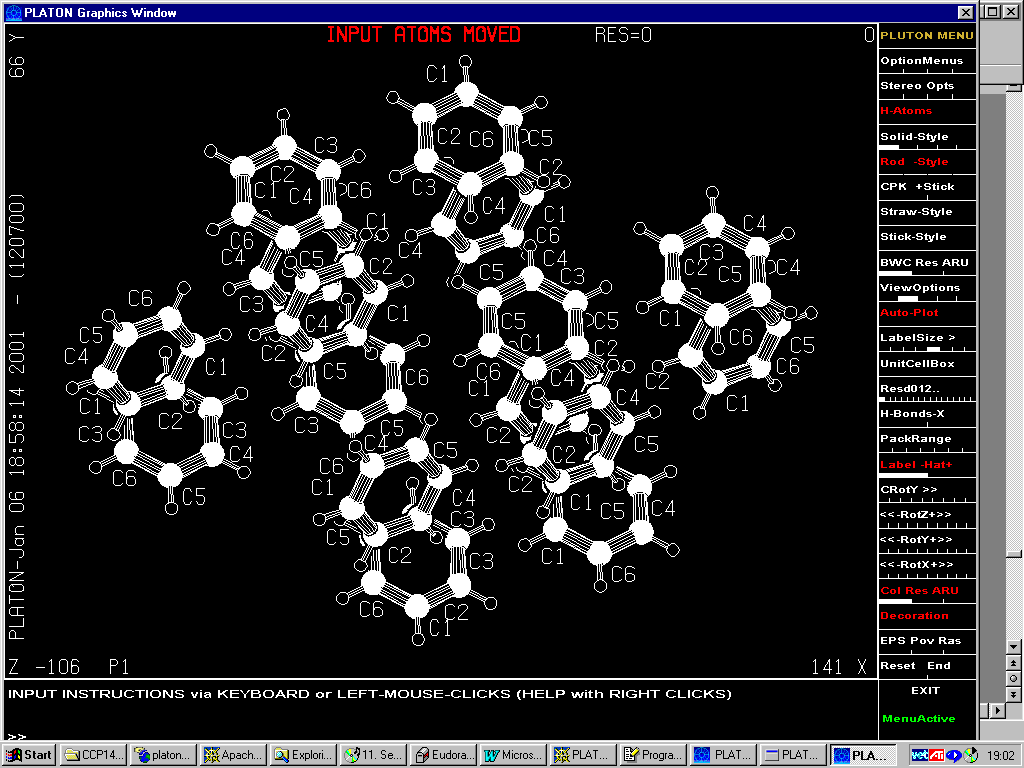
Using < 20th Nov 2000 Platon/Addsym
Gives P –1
(1/2 subcell)(24 independent Carbon atoms)
7.8517 9.7483 12.7427 87.496 80.494 77.047
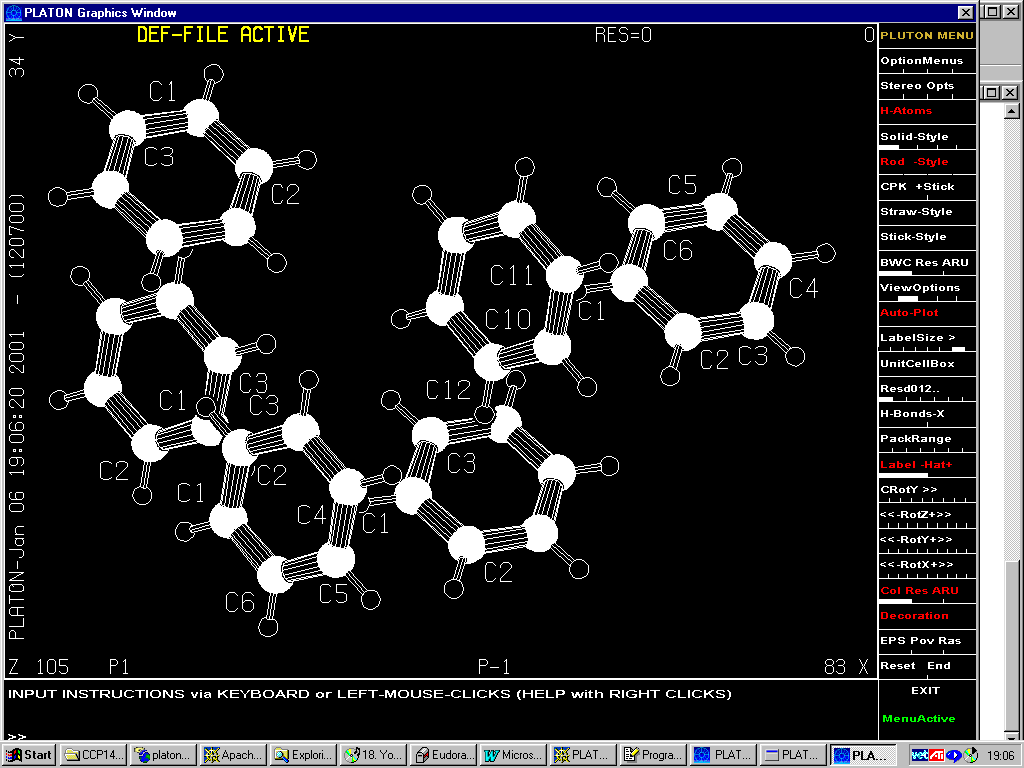
Using => 20th Nov 2000 Platon/Addsym
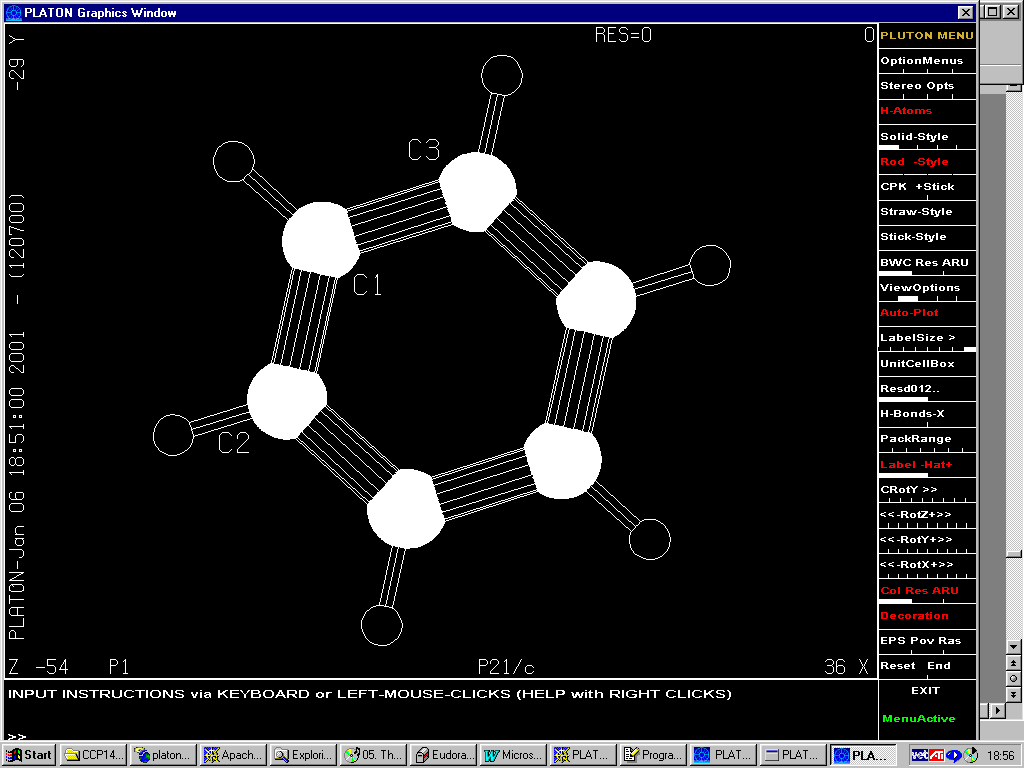
Gives P21/c
(1/8 subcell !)(3 independent Carbon atoms)
5.5375 5.5660 8.0032 90.000 108.177 90.000
Specialist Applications
(Single Crystal and Powder Diffraction)
Charge Density (single crystal)
Project XD
http://www.chem.gla.ac.uk/~paul/paul.html
Anharmonic Refinement
List of Software:
http://www.ccp14.ac.uk/solution/anharmonic/
Incommensurate Structure Refinement
List of Software:
http://www.ccp14.ac.uk/solution/incomm.htm
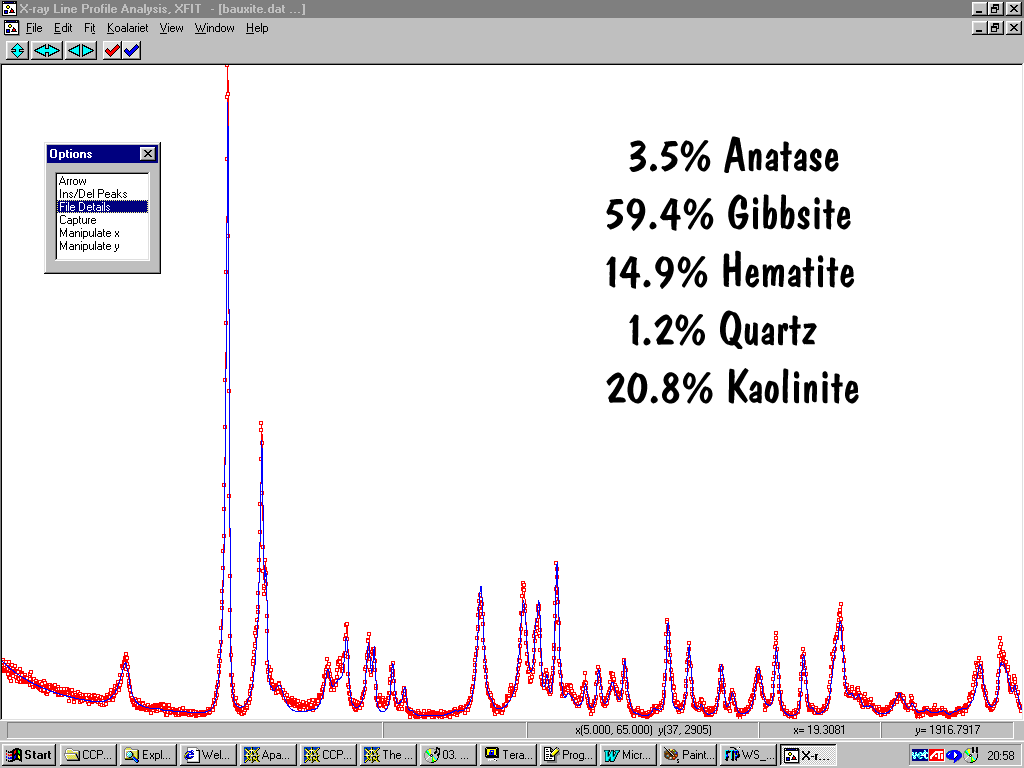
Quantitative Phase Analysis
Non-trivial and in many cases, custom solutions may be required. (a complete symposium in itself)Rietveld programs are commonly used for Quantitative Analysis (refer above list).
Maud for Java Quantitative Analysis Tutorial:
http://www.ing.unitn.it/~luttero/maud/tutorial/
Refer to non-Rietveld references cited in:
http://www.ccp14.ac.uk/poster-talks/david-hay-quant-notes-axaa99/
Powder Diffraction Pattern Calculation
Most Rietveld Programs can calculate powder patterns but may not be all that friendly to use compared to the following dedicated software:
Powder Cell
http://www.bam.de/a_v/v_1/powder/e_cell.html
Imports common file formats and GUI based editing of structure:
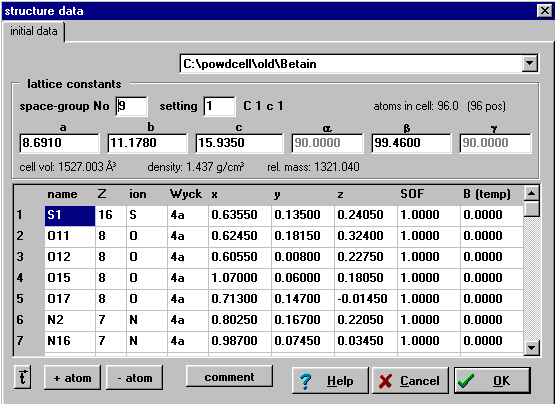
GUI based manipulation of structure as well as powder pattern.
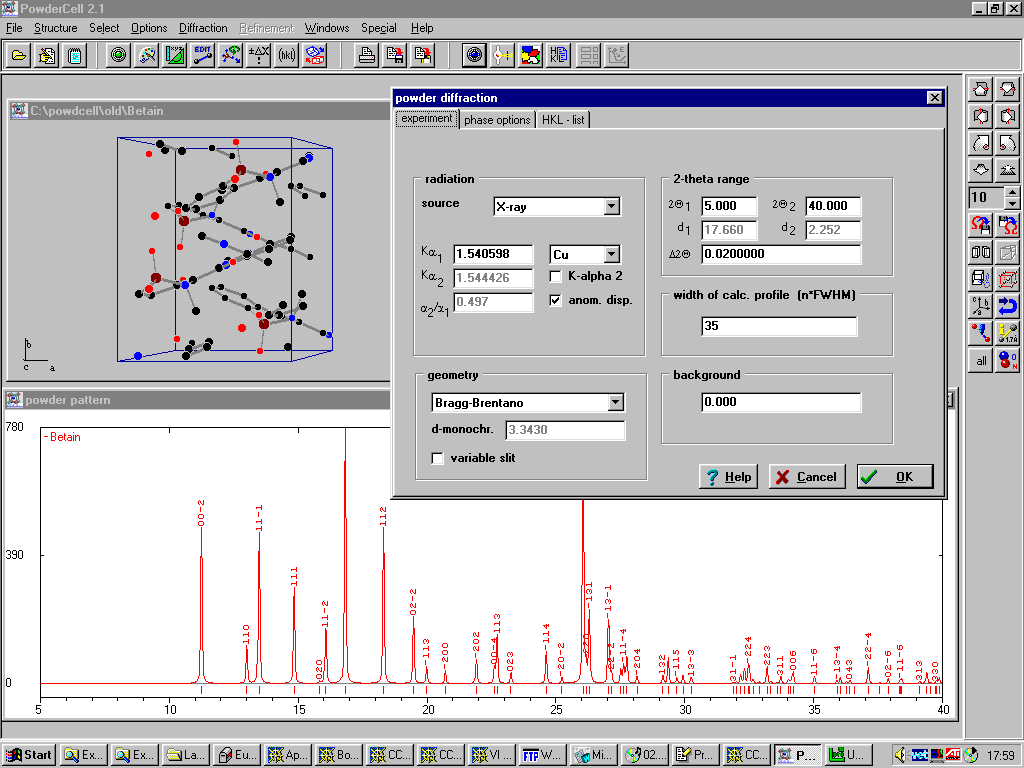
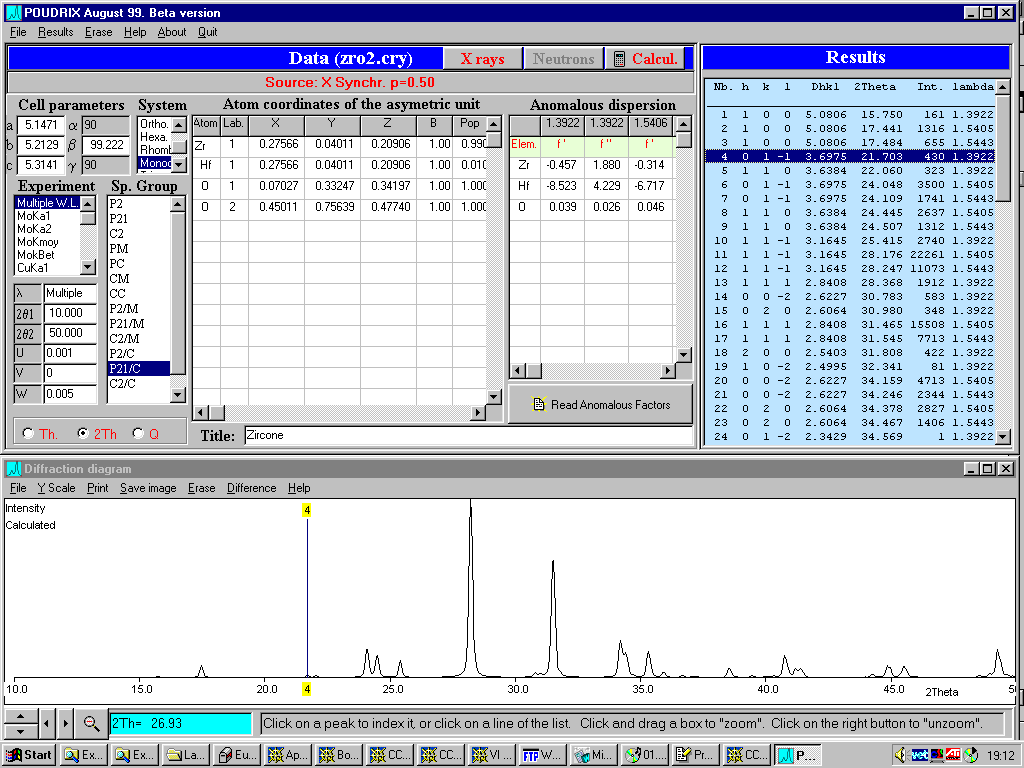
Poudrix
http://www.ccp14.ac.uk/ccp/web-mirrors/lmgp-laugier-bochu/
Imports common file formats and GUI based editing of structure:
Can accurately model Anomalous Dispersion for Synchrotron X-rays.
Structure Visualisation and Photorealistic Hardcopy Output
Examples of Software that can do this:
(problem can be that or having a high enough quality printer)
ORTEX (Images and Movie Animations):
http://www.nuigalway.ie/cryst/
http://www.ccp14.ac.uk/ccp/web-mirrors/ortex/cryst/
Just open up a Shelx format *.INS/*.RES file and go for it
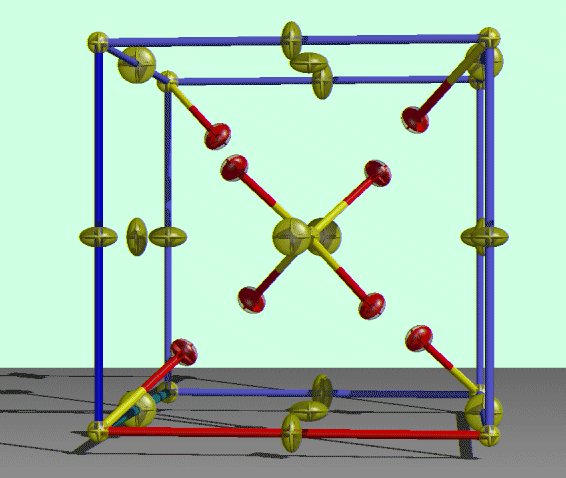
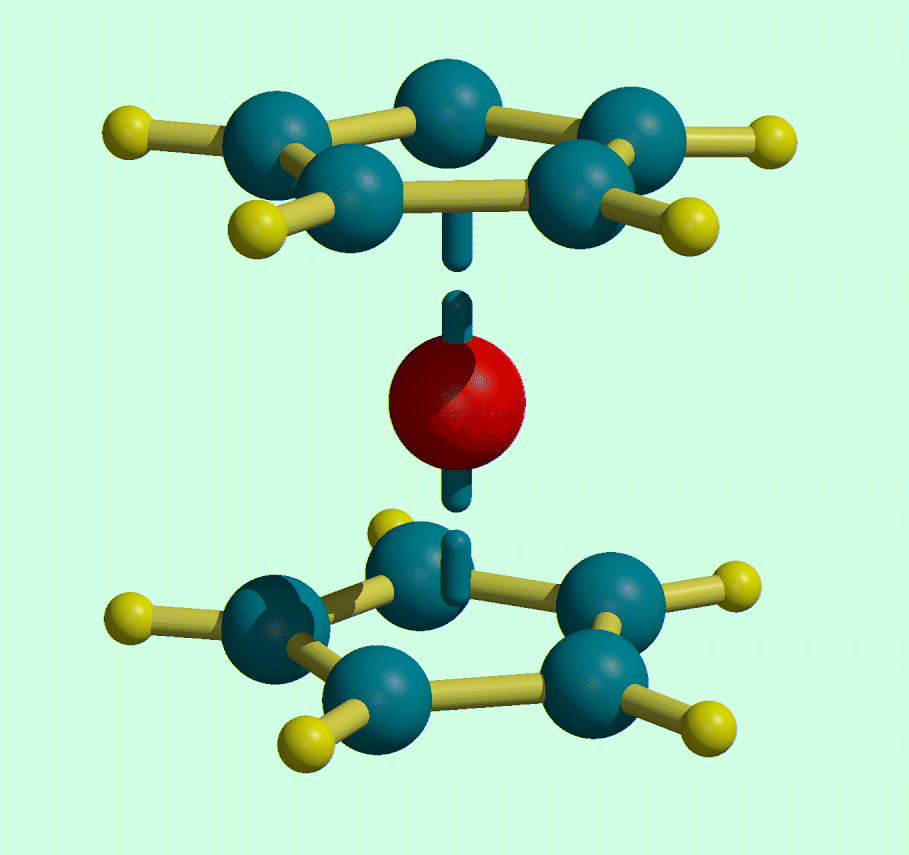
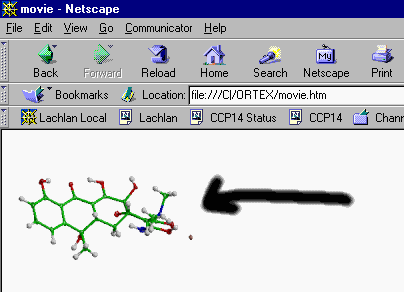
Movie Making in ORTEX
Open a Shelx File and optimise the view in ORTEX
Start the RASMOV Movie Generator
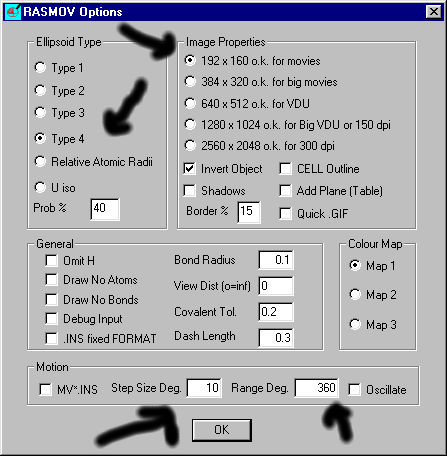 Set the Options you Desire:
Set the Options you Desire:
Web Ready Animated GIF will be generated:
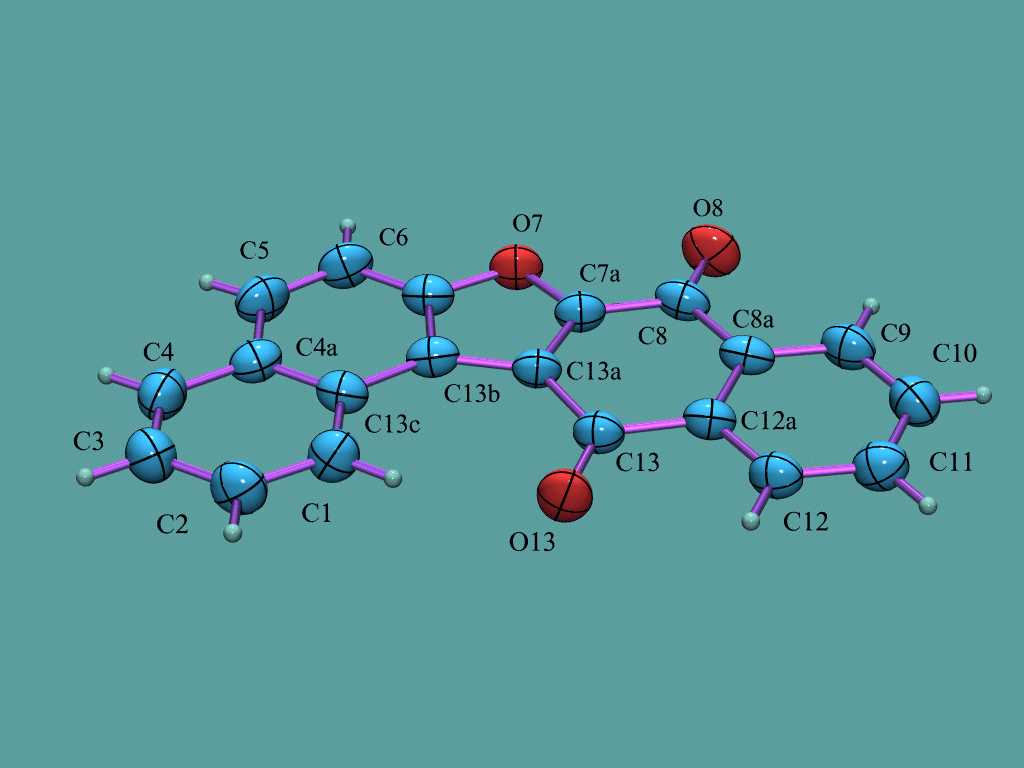
GUI WinOrtep 3/WUI WinStruplo/WinGX
http://www.chem.gla.ac.uk/~louis/software/
http://www.ccp14.ac.uk/ccp/web-mirrors/farrugia/~louis/software/
Can load a variety of structure formats including some Rietveld formats such as GSAS, LHPM, Fullprof, Rietan.
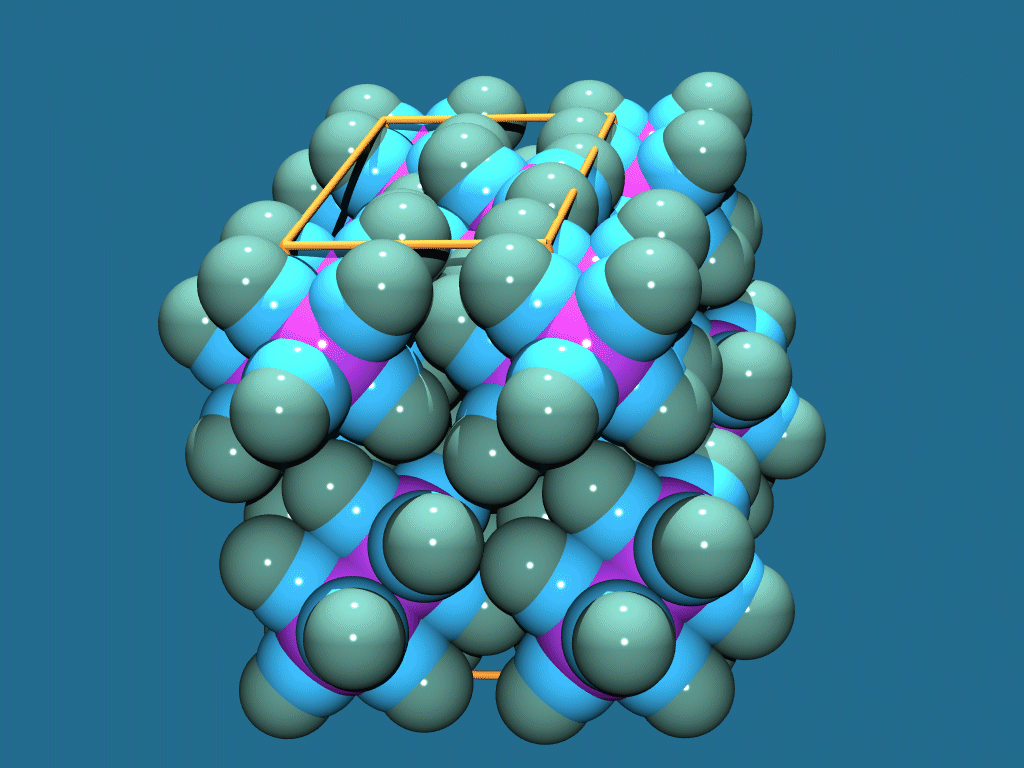
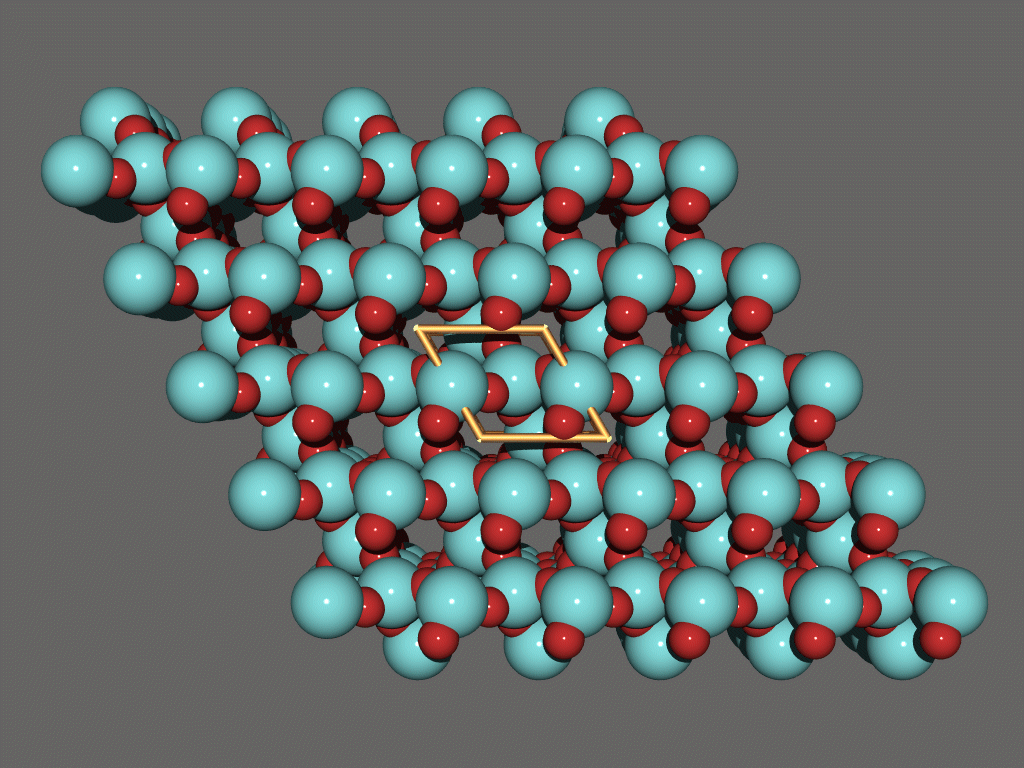

Platon/Pluton/System S
http://www.cryst.chem.uu.nl/platon/
http://www.ccp14.ac.uk/ccp/web-mirrors/platon-spek/platon/
GNU Xtal Suite
http://xtal.crystal.uwa.edu.au/
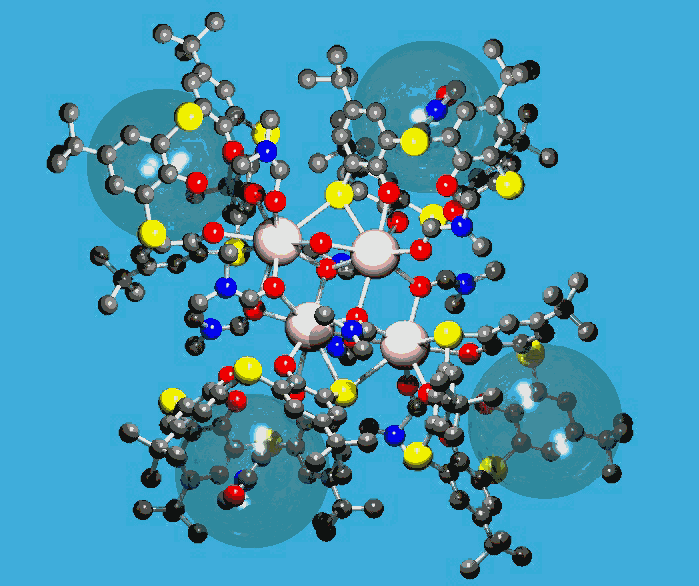 http://www.ccp14.ac.uk/ccp/web-mirrors/xtal/
http://www.ccp14.ac.uk/ccp/web-mirrors/xtal/
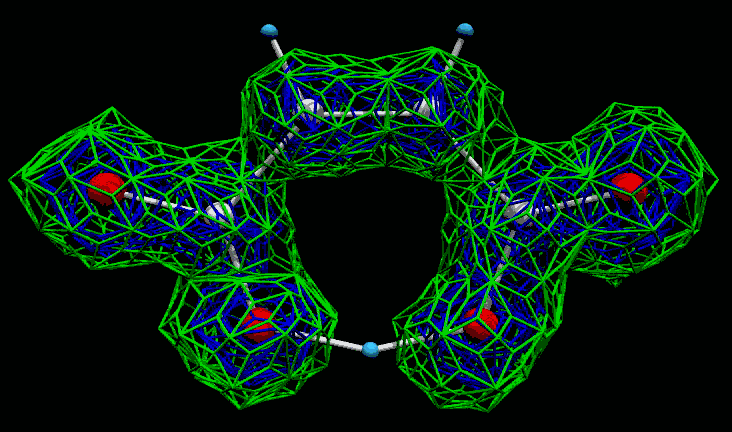
Marching Cubes 3D Fourier Map Viewer
(links into Crystals and WinGX. GSAS via FOUE) –
outputs Povray files
http://mysak.umbr.cas.cz/~husakm/Public/MarchingCubeELD/MarchingCubeELD.htm
http://www.ccp14.ac.uk/ccp/web-mirrors/marchingcube-fourierviewer/~husakm/Public/MarchingCubeELD/MarchingCubeELD.htm
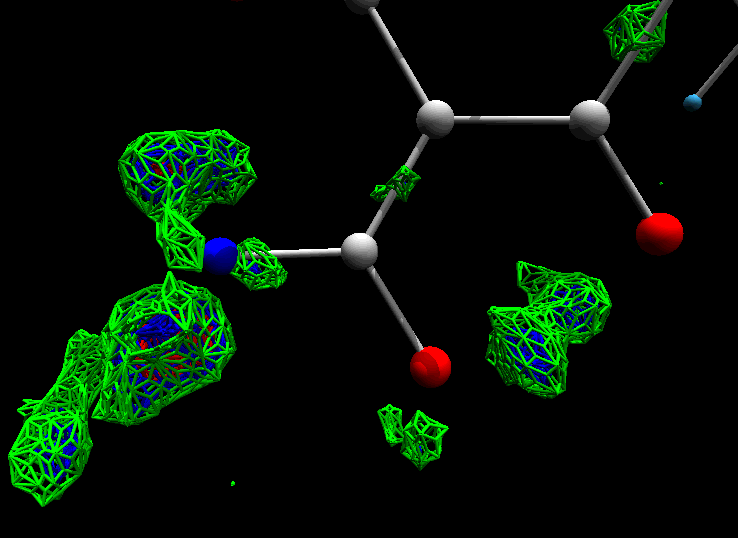
Dual Boot UNIX/Windows PCs
Obtain the ability to run a very wide range of crystallographic software on cheap, powerful commodity PCs.
Tutorials exist for helping you do this:
CCP14 based tutorials on installing multi-boot
A local UNIX advocate would be quite happy to get a dual boot system going for you.
Free UNIX Operating Systems (install off the internet)
Linux
refer:
http://www.ccp14.ac.uk/solution/linux/FreeBSD (can run linux binaries)
refer:
http://www.ccp14.ac.uk/solution/bsdunix/(be careful of hackers invading your systems when running Linux/UNIX. CCP14 tutorials try to be security conscious)
VMWARE (simultaneously run of Windows and Linux):
http://www.vmware.com/
(Commercial software)
Freely Available Compilers, GUI Building Kits and Source Code
http://www.ccp14.ac.uk/recomm/
If Isolated from the Internet (or on very slow links)
Free
Xtal Nexus CD-ROMs for academics and studentshttp://www.unige.ch/crystal/stxnews/nexus/index.htm
(Supported & Sponsored by the IUCr, ICSU and CCP14)

Contact: Lachlan Cranswick
CCP14
CLRC Daresbury Synchrotron Laboratory,
Warrington, Cheshire, WA4 4AD U.K
E-mail: l.cranswick@dl.ac.uk
WWW: http://www.ccp14.ac.uk
Summary
Downloadable via the EPSRC funded CCP14 website:
http://www.ccp14.ac.uk
E-mail: ccp14@dl.ac.uk