An Inorganic Crystal Structures Generator
Reference
Package
New
Starting
The .dat file
Satellites
GRINS
Output files
NEW : version for dual-core processors
GRINSP = Geometrically Restrained INorganic Structure Prediction
GRINSP is a Monte Carlo code (FORTRAN)
for the prediction of
inorganic crystal structures built
up from defined polyhedra.
Version 2.00 works with any standard
space group, building models with
3-, 4-, 5- and 6- vertices
polyhedra connected exclusively by
corners, single polyhedra or binary.
More to come, perhaps...
Last modification : October 2006
No wizardry with GRINSP predictions,
only a cute (?) algorithm.
Introduction
The concept of inorganic crystal structure prediction by using geometrical restraints is not new :
- Zeolite researchers have documented more
than 1000 hypothetical structures by using classical physical model building
[1] during the past 60 years.
- Simulated annealing is a rapid generator
of hypothetical 4-connected framework structures and others. More than
5000 hypothetical zeolite structures were reported in ref [2]. More than
1.000.000 are now in the Hypothetical
Zeolites Database.
- Many recent works in inorganic structure
prediction (as well as organic and organometallic) have produced huge quantities
of hypothetical compounds (using commercial packages as CERIUS, etc), no
room here for citing them all.
- Systematic enumeration is now based
on advances in mathematical tiling theory [3-4].
Where are these predicted structures ? A few of them are inside of the ICSD, for instance some theoretical SiO2 structures [5]. More than 1.000.000 zeolite models are inside of the Hypothetical Zeolites Database. But what about predicted compounds other than SiO2 ? The main purpose of GRINSP(*) is to generate hypothetical inorganic structures MxM'yXz which will be documented in a searchable database : PCOD (Predicted Crystallography Open Database), a subset of the COD. If you are a good "predictor" and want to deposit your predicted structures in PCOD, this is already possible (CIF files only), visit the upload page.
GRINSP does not work by applying simulated annealing to a starting random configuration. Version 2.00 works schematically as follows, by using the Monte Carlo method :
- Manual selection of the constraints on cell parameters, of restrained interatomic distances, of the type(s) of coordinations, and of the space groups. Then the Monte Carlo process starts.
- Random selection of the cell parameters inside of the predefined range.
- Random positioning of a first cation M (or M') of the future MxXy (or MxM'yXz) compound on a general or special position, itself selected randomly.
- Random positioning of the next cations (random choice of M or M') in respect of the distance restraints with the atoms already accepted, on a general or special position, itself selected randomly.
- If a model fulfills all distance and coordination criteria, place the X atoms at M-M midpoints, refine the atomic positions and cell parameters so as to improve an R factor.
- Continue to try to predict structures in that way till a certain number of cells are tested.
- Find if the predicted structures are new or were already described (using CS - Coordination Sequences), keep those with best R factors.
Currently, there are some limitations in GRINSP 2.00 which proved to be efficient for a maximum number of 192 M/M' atoms on up to 1 to 6 different general or special positions. It was shown to be able to predict many zeolites (ABW, ACO, AFI, BIK, EDI, FAU, GIS, LTA, SOD... but not all of them) or the compact SiO2 phases (quartz, cristobalite, tridymite, etc), polymorphs for B2O3, MF3 (M = Al, Fe, Cr...), hypothetical phases in binary systems B2O3/SiO2, B2O3/ReO3, SiO2/ReO3, titanosilicates, etc (see the PCOD). It is up to you to try GRINSP with other systems, and even the above ones have not been completely explored.
Further work is needed for improving the GRINSP efficiency :
- Introduction of different linkage modes than by corners (edges, faces...)
- Adding the possibility for insertion of big cations K/Sr/Ba/Cs/etc as spheres in the holes/tunnels
- Considering bond valence as an alternative to pure geometrical restraints for the model final refinements
- Increase speed by not recalculating always everything (distances)
- Increase the box size for the CS (coordination sequence) calculations (729 cells is not always enough...)
- Make a parallel version, working on dual-core processor or biggest multi-processor machines
- Produce a database of calculated powder patterns which would allow early identification of new phases and even structure solution before indexing or in spite ot non-indexation
- Etc !
[1] J.V. Smith, Chem. Rev. 88 (1988) 149-182.
[2] M.W. Deem and J.M. Newsam, J. Am.
Chem. Soc. 114 (1992) 7189-7198.
[3] O. Delgado Friedrichs, A.W.M. Dress,
D.H. Huson,, J. Klinowski, A.L. Mackay, Nature 400 (1999) 644.
[4] M.D. Foster, O. Delgado Friedrichs,
R.G. Bell, F.A. Almeida Paz, J. Klinowski, Angew Chem. Int. Ed. 42 (2003)
3896-3899.
[5] M.B. jr Boisen, G.V. Gibbs, M.S.T.
Bukowinski, Phys. & Chem. of Minerals (Germany) 21 (1994) 269-284.
(*) If you find "GRINSP" unpronounceable,
suggest another name to alb@cristal.org
, other possible names, not retained, were "INORGOD" (only God can predict...)
or "INORGURU", "AUGUR", "PREDINORG"...
Using GRINSP, you should cite:
"Inorganic Structure Prediction with
GRINSP"
A. Le Bail
J. Appl. Cryst., 38 (2005) 389-395.
The corresponding PDF
is available on this Web site
and at the IUCr (Open Access)
J. Appl. Cryst.38,
389---395
Older texts are still available. An introduction to GRINSP and PCOD was published in the CPD Newsletter 31, a longer paper was published in the IUCr Computing Commission Newsletter (July 2004).
The most recent powerpoint presentations about GRINSP, PCOD (and COD) were made at the IUCr XXth meeting, Florence (August 2005). Files are available.
Another text about hypothetical AlF3 crystal structures is available :
A. Le Bail & F. Calvayrac,
J. Solid State Chem. 179 (2006) 3159-3166.
http://dx.doi.org/10.1016/j.jssc.2006.06.010
finally, predicted titanosilicates were presented at EPDIC 10.
Packages
GRINSP is distributed in two packages :
- grinsp.zip version for monoprocessor
- pgrinsp.zip version for dual-core processors (see details below)
Both packages contain, in appropriate
directories :
- grinsp.exe : GRINSP version 2.00, executable for Win95/98/NT/XP.
- grinsp.pdf : a copy of the text published in the J. Appl. Cryst. (2005).
- grins.zip: contains the GRINS satellite program for model optimization after cation/anion substitution.
- cutcifp.zip : contains the CUTCIFP satellite program for cutting multiple CIFs.
- cif2con.zip : contains the CIF2CON satellite program reading a multiple CIF and creating a .con file with coordination sequences
- connect.zip : contains the CONNECT satellite program for analysis of coordination sequences (.con file)
- framdens.zip : contains the FRAMDENS satellite program for listing the compounds with smallest densities.
- index.zip : Containing this help file, index.html, and additional image files.
- connectivity.txt : The coordination sequences (CS) of known zeolites and dense SiO2.
- distgrinsp.txt : file containing the geometrical restraints for a few given atom pairs (you may add your own data there).
- wyckoff.txt : file containing the general and special position codes for all standard space groups. Note that the true multiplicity is regenerated by GRINSP after application of the Bravais translations.
- examples.zip : Some example files.
- grinsp.ico : an icon for GRINSP, a few tetrahedra coming out from a wizardry hat.
- the pgrinsp package contains an additional file : libguide40.dll, to be installed in the same directory as grinsp.exe
Unzip all these files in any directory named at your convenience, and run the program (no DLL needed, nothing to change in the autoexec.bat file...). Note that connectivity.txt, distgrinsp.txt and wyckoff.txt have to be absolutely in the same directory as grinsp.exe and the .dat file defining your expected predictions.
The source codes
The FORTRAN source codes for GRINSP and the satellite programs (GRINS, etc) ready for compilation ("console application") by the Intel Visual Fortran 9.1 compiler can be found into the .zip files, with .f extension.
Example files
sio2.dat : File ready for the prediction
of zeolites by GRINSP.
titanosilicates.dat : File ready
for the prediction of titanosilicates by GRINSP.
test129.dat : File for the prediction
of t-AlF3 in the P4/nmm space group by GRINSP.
GaF3.dat and TiP.dat : Files ready
for GRINS.
Examples.zip contains also the .imp files, if you wish to compare with your results (note that this is Monte Carlo, you may obtain the same models in a different sequence).
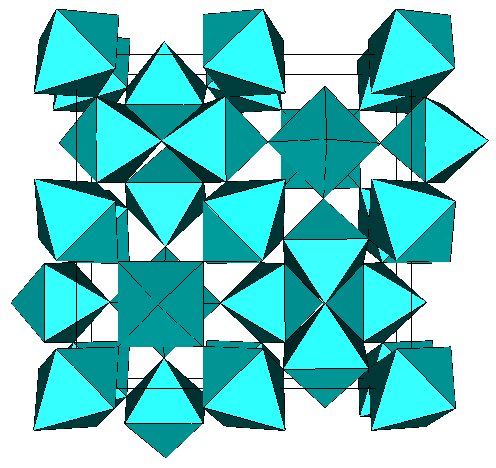
Below is the best output for test129.dat
the t-AlF3 6-connected 3D network :
October 2006 : PGRINSP package
- GRINSP inside of the PGRINSP package is the version for parallel computing able to exploit dual-core processors (tested only with Intel Pentium D...), using OpenMP directives as available inside of the Intel Visual Fortran compiler 9.1.
- You will note some changes in the order of the solutions displayed on the screenbox during the run. If say ncells =10000 cell tests are required per space group, one processor will work on ncells = 1 to 5000 and the other will work on ncells = 5001 to 10000. Otherwise, no change in datafile or output files.
- Speed : between 1.7 and 1.8 times faster with 2 processors than with only one.
- Quad cores in 2007 ;-)
- 80 cores in 5 years ???
Trick : if you cannot work well with your PC when GRINSP is running, decrease the GRINSP priority to lower than normal. This is done with the task administrator (Ctrl Alt Supp). Go to "process", select the GRINSP process with the mouse right button, select "priority" and decrese it.
NOTE : The libguide40.dll
has to be installed in the same directory as grinsp.exe
April 2006 : Improvements in GRINSP version 2, if compared to version 1.
- Larger models can be predicted (limit
now at 192 M/M' atoms instead of 64). Structures as
complex as faujasite can be produced.
- Bugs corrected (the coordination sequences are no longer giving strange results)
- More user-friendly, parameter file simplified
:
+
a range of space groups can be examined inside of the same run, instead
of only one,
+
space groups are specified by their number instead of the Hermann-Mauguin
description
+
no range angles to provide
- More details inside of the output CIFs
:
+ better
analysis of the formula and Z,
+ output
of the M/M' starting Wyckoff positions (before optimization) so that retrieving
the
true space group from the P1 description is facilitated,
+ output
of the FD (Framework Density).
- GRINS allowing for the computation of
isostructural compounds is considerably improve, and
can read multiple CIFs of a previous series from GRINSP : titanosilicates,
once
modelled can lead fast to isostructural titanophosphates, vanadophosphates,
gallophosphates, etc.
- Satellite programs are provided for global
analysis of the (sometimes) huge lists of predicted
structures:
+ CUTCIFP
can read a multiple CIF and provide single CIFs having names changes at
your
convenience,
+ CIF2CON
can read a multiple CIF and provide a .con file containing the connectivity
sequences (CS) for further use and identification of unique models,
+ CONNECT
can read a .con file, identify unique models, compare to a previous list
of
models characterized by their CS (stored into the file connectivity.txt),
and provide a sorted
classification by decreasing order of the R factor.
+ FRAMDENS
reads a multiple CIF and produces the list of compounds ordered according
to their framework densities (number of cations for 1000 A3),
allowing to point at the best
models with largest tunnels/cavities.
Running the Program
Verify first that the working directory contains grinsp.exe, connectivity.txt, distgrinsp.txt, wyckoff.txt and your parameter file with .dat extension (for instance SiO2.dat as below). The Windows PC version will run by clicking on grinsp.exe, opening a window shown below :
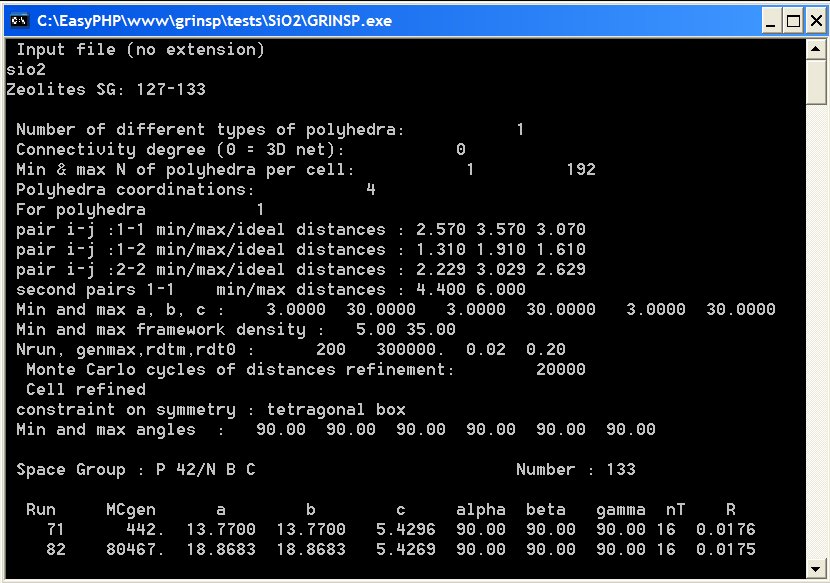
select your data file, for instance SiO2.dat
from the example files (do not type the .dat extension). The entry data
parameters are displayed, as well as a summary for every successful prediction.
You
may stop the program execution by typing K (capital letter) anytime.
The program will store and sort the results, and stop at the next multiple
of 50 runs (so the stop is not immediate but may need a few seconds), See
now what are exactly the parameters into the starting .dat file:
Some changes were made in GRINSP version
2.00.
An example (SiO2.dat) is detailed below
:
Zeolites SG: 16-74 : text for this run 16 74 : SG : Space groups range (two values between 1 230) 1 0 1 192 : npol, ncon, nmim, nmax 4 : ncpol (coordination for each polyhedron-type) Si O : elements for a search in the distgrinsp.txt file 3. 30. 3. 30. 3. 30. : min and max cell parameters a, b, c 5. 35. : min and max framework density (FD) 1000 300000 0.02 0.12 : ncells, genmax, Rmax, Rdt0 6000 1 : idls (MC cycles for distance refinement) and iref (1 or 0) 1 : code for selecting models to output
note the main differences :
Titanosilicates - SG: 188-194 188 194 2 0 2 192 npol = 2 here instead of 1. 6 4 note the two coordinations defined here (octahedron and tetrahedron) Ti O one line for the TiO6 octahedron Si O and one line for the SiO4 tetrahedron 3. 30. 3. 30. 3. 30. 5. 35. 2000 300000 0.02 0.12 6000 1 1
text A title for the prediction session, 80 characters max (format 20A4).
SG
The space group(s) range examined (two integers : SG numbers)
examples :
1 230 : all space groups examined starting
at 230, decreasing symmetry.
74 74 : only one space group examined
: N°74
195 230 : cubic space groups examined stating at 230, decreasing
to 195
GRINSP contains all standard space groups
(if you want non-standard SG, add them in Wyckoff.txt
npol, ncon, nmim, nmax : four integers (free format)
npol = number of different types
of polyhedra you wish in the predicted structure
Warning : only
npol = 1 or npol = 2 are allowed in GRINSP version 2.00.
ncon = defines
the degree of connection between polyhedra
0 : every X atom is connected to two M/M' atoms (corner-sharing)
> 0 : some X atoms are connected to only one polyhedron
< 0 : there could be edge-sharing, etc
Warning : only
ncon = 0 is working in GRINSP version 2.00
nmin =
minimal mumber of M/M' atoms for saving a model
(if you are tired to see these small stuctures with nT = 1, 2, 3 or 4 all
the time)
For exploring a binary system, only solutions mixing M and M' will be retained
(meaning that the minimum will be nmin=2, anyway)
nmax =
maximal mumber of M/M' atoms for saving a model (max = 192)
if you are exploring small cell volumes, reduce nmax to appropriate values
(20, etc)
this will save computer time (avoiding to test Wyckoff positions corresponding
to
too much atoms).
ncpol = coordination
for every of the npol different polyhedron-type
Warning ! only
four values ncpol = 3, 4, 5 or 6 are allowed in GRINSP version 1.00
and a maximum
of 1 or 2 values can be given (because npol = 1 or 2 maximum)
Elements : two elements for a given polyhedron - there may be two lines if a binary system is explored. This part is formatted as 2A4 : four cases per atom.
GRINSP will have to find the minimal/maximal/ideal
interatomic distances for this polyhedron inside of the file distgrinsp.txt
After the same atom codes in distgrinsp.txt
is added the coordination, and then 4 lines corresponding to the prescribed
interatomic distances :
Si O 4
: atom codes and coordination (here a SiO4 tetrahedron)
2.60 3.60 3.070 : minimal/maximal/ideal first distance
between atom pair 1-1
1.30 1.90 1.610 : minimal/maximal/ideal first distance
between atom pair 1-2
2.20 3.00 2.629 : minimal/maximal/ideal first distance
between atom pair 2-2
4.40 6.00
: minimal/maximal second distance between atom pairs 1-1
You may edit distgrinsp.txt and add there your own data (anywhere) : 5 lines per kind of polyhedron as above. Care that the elements are given in 2A4 format.
cell = one line
giving the cell parameter ranges to explore for structure prediction
6 values : amin, amax, bmin, bmax, cmin, cmax
free format
Framework density,
min and max.
The framework
density is the number of M/M' atoms for a volume of 1000A3.
Solutions outside
of this given range will be excluded. This allows to avoid retaining some
two-dimensionnal
crystal structures, if you do not want them. Be careful to allow a sufficiently
large range (
5 to 35 should be correct for zeolites)
ncells = number of different cells examined per space group (use 200-20000) There is no maximum in fact, but testing 1000 cells may need 10 to 60 minutes. These cells are proposed randomly by the Monte Carlo process.
genmax = number of Monte Carlo trials for a given cell use 10000-500000, the latter being time consuming... Rmax = the structure predictions corresponding to the R factor lower than Rmax will be stored and sorted (use Rmax = 0.005-0.01 if you wish regular polyhedra, up to 0.02 if you tolerate distortion, up to 0.03-0.05 if you expect trigonal prisms instead of octahedra, or even pentagonal pyramids, etc) The R value is analogous to the RDLS (see the Database of Zeolites structures), but is obtained by Monte Carlo, not by a least-squared refinement. Use no more than Rmax = 0.02 if you wish to upload your predicted structure into the PCOD. Rdt0 = the candidate structures corresponding to the R factor lower than Rdt0 before optimization will be optimized (use Rdt0 = 0.10-0.20). The more Rdt0 is small, the more the optimizations have chances of being successful (leading to R < Rmax). In general, one third to half of the candidate structures have Rdt0 < 0.13 before optimization. Some statistics are given at the end of the .imp file. See the study about speed below.
Note that from genmax are calculated 'insistence factors' (IF). These IF are 6 values corresponding to the number of Monte Carlo trials during which to insist before to change some parameters (selecting a new cation for completing its environment, or selecting which new coordination for a new cation to add, or etc) : - value 1 is for insisting on placing a second atom - value 2 is for insisting on the completion of the cationic neighbouring of a cation having already one previous neighbour - value 3 is for insisting on the completion of the cationic neighbouring of a cation having already two previous neighbours - value 4 is for insisting on the completion of the cationic neighbouring of a cation having already three previous neighbours - value 5 is for insisting on the completion of the cationic neighbouring of a cation having already four previous neighbours - value 6 is for insisting on the completion of the cationic neighbouring of a cation having already five previous neighbours These insistence factors are fixed to be equal to IF1=genmax/320, IF2=genmax/160, IF3=genmax/80, IF4=genmax/40, IF5=genmax/20 and IF6=genmax/10 if the maximal coordination is larger than 4, and to be equal to IF1=genmax/80, IF2=genmax/40, IF3=genmax/20 and IF4=genmax/10 if the maximal coordination is smaller or equal to 4. So that the choice of genmax may be critical. The author uses generally genmax = 300000.idls, iref
idls = number of Monte Carlo steps for the interatomic distances and cell improvement (use 20000) at the optimization stage
iref = code for cell improvement (iref=1) or not (iref=0) if iref = ,1, half of the idls Monte Carlo steps wll be used for the cell improvement.output code
1 or -1
if 1 : output of new solutions, having Coordination Sequences not already existing into connectivity.txt, or having a better R. if -1 : output of all solutions (not only those being new).
GENERAL LIMITS: 10000 optimized structures (2000 in PGRINSP) 192 M/M' cations 5000 coordination sequence in connectivity.txt (2000 in PGRINSP) 576 anions 2 kinds of polyhedra
Satellite Software
GRINS
This satellite program can optimize previous GRINSP models, changing the M/X and/or M'/X couples (building isostructural compounds).
The organization of the .dat file is much simpler than for GRINSP. Details for the model to be transformed are obtained from a CIF previously built by GRINSP (the model description should be exclusively in P1 space group). GRINS can also cope with multiple CIFs, transforming for instance a long series of titanosilicates into gallophosphates (etc).
Two examples are below with the GaF3.dat (Ga replacing Fe) and TiP.dat (Ti and P replacing Ti and Si) files :
GaF3.dat file :
Test : building GaF3 from FeF3 FeF3 ! the filename of the CIF containing previous model(s) 1 ! number of different polyhedra 6 ! coordination(s) of the polyhedra Fe F ! couple M/X in the previous model(s) Ga F ! new couple M/X for the isostructral compound 5 ! nruns : number of different tests (for finding the best) 5000 1 3 ! optimization steps, cell refined, lines in the original CIF 3300000 ! first filenameTiP.dat file :
Test : building titanophosphates from titanosilicates total-cif-best ! the filename of the CIF containing previous model(s) 2 ! number of different polyhedra 6 4 ! coordination(s) of the polyhedra Ti O ! couple M/X in the previous model(s) Si O ! second couple in the previous model(s) Ti O ! new couple M/X for the isostructral compound P O ! second couple for the isostructural compound 5 ! nruns : number of different tests (for finding the best) 5000 1 2 ! optimization steps, cell refined, lines in the original CIF 2201000 ! first filename
The parameter "line" above corresponds to the number of lines between the line containing "Probable space group" and the line starting by _cell_length_a in the CIF containing the model(s). In the example below, there are 3 lines. Some old results from GRINSP version 1 had not the cell formula unit, so that there would be only 2 lines.
data_PCOD2201011 _publ_section_title ; Structure prediction by GRINSP 2.00 - 2006 (A. Le Bail) TiPO5 PCOD2201011 R = 0.0051 Probable space group: P -4 21 C ; _chemical_formula_sum "Ti P O5" _cell_formula_units_Z 4 _cell_length_a 6.4086 _cell_length_b 6.4086 _cell_length_c 7.7859 _cell_angle_alpha 90.000 _cell_angle_beta 90.000 _cell_angle_gamma 90.000 _cell_volume 319.77GRINS places the X atoms at the (M/M')-(M/M') midpoints and executes the optimization process. It is advisable to make 5-20 tests per model (this is Monte Carlo). The models with minimum R will be listed in the .imp file.
Note that the following files have to be placed
into the
same directory as grin.exe, the .dat file
and the previous models (.cif) file :
connectivity.txt
distgrinsp.txt
wyckoff.txt
CUTCIFP
Satellite software allowing to cut a multiple CIF into individual CIFs, changing all PCOD numbers at your convenience. Note that the building of multiple CIFs can be made by concatenation of single CIFs by using the command line :
C:\directory\copy *.cif total.cif
In the example delivered into cutcifp.zip in the package, all the 89 CIFs produced by the SiO2.dat example were concatenated, then they were cutted by CUTCIFP with a change of their PCOD numbers in a continuous series from PCOD4500000 to PCOD4500088, then all these individual CIFs were again concatenated into only one multiple CIF that was then processed by CIF2CON below (use numbers with 7 digits).
CIF2CON
Satellite software allowing to extract all the coordination sequences (CS) from a multiple CIF. The created file is names with the .con extension.
In the example delivered into cif2con.zip, the multiple CIF file total.cif is processed by CIF2CON, giving the starting file number 4500000, producing a file named total.con. A part of the content of that file is shown below :
PCOD4500000 SiO2 R = 0.0092 19 P 21 21 21 filenumber plus some details (formula, R, SG number 2 number of different nodes 8 4 number of equivalent atoms for each node 4 12 30 48 76 114 152 196 0 0 connectivity sequence for each node 4 12 28 50 80 110 152 198 0 0 PCOD4500001 SiO2 R = 0.0083 19 P 21 21 21 4 4 4 4 4 4 11 26 43 68 101 132 174 221 267 4 11 25 44 69 97 135 172 218 275 4 11 24 43 68 98 134 173 222 265 4 11 23 44 69 94 131 181 213 277 PCOD4500002 SiO2 R = 0.0090 19 P 21 21 21 1 12 4 12 26 46 70 100 136 178 0 0
independently of the cells, space groups or atoms.
CONNECT
Satellite software allowing to analyze a .con file built up by using CIF2CON, and to detect the models having the same coordination sequences (CS). A list of the best models (with best R values) is proposed at the end of the result file with .txt extension.
In the example delivered into connect.zip, the file total.con is analyzed by running CONNECT. Note that the connectivity.txt file is required. In that case, it contains all the connectivity sequences of the known zeolites. The 89 CIFs produced by GRINSP with the test case SiO2.dat are shown by CONNECT to be reduced into 48 distinct models. CONNECT produces the result into the file named total.txt. At the beginning of that file, are listed the 89 first lines of the connectivity sequences found into total.con. Then the analysis starts by comparison of the connectivity sequences :
PCOD4500000 is probably new PCOD4500001 is probably new PCOD4500002 is probably new PCOD4500003 is probably new PCOD4500004 is probably new PCOD4500005 is probably new PCOD4500006 is probably new PCOD4500007 is probably PCOD4500005 R factors 7.8999996E-03 9.7000003E-03 but with better R, or =... 7.8999996E-03 9.7000003E-03 update made PCOD4500008 is probably new PCOD4500009 is probably PCOD4500008 R factors 6.3000000E-03 6.6999998E-03 but with better R, or =... 6.3000000E-03 6.6999998E-03 update made EtcAt the end of the file total.txt is provided the list of best individual models (with lowest R factors):
models to save : 48 Models ordered according to R PCOD4500051 SiO2 R = 0.0027 44 I M M 2 one model PCOD4500057 SiO2 R = 0.0034 45 I B A 2 two times the same model PCOD4500056 SiO2 R = 0.0035 45 I B A 2 note that next models are beginning by a space PCOD4500050 SiO2 R = 0.0040 43 F D D 2 4 times the same model PCOD4500047 SiO2 R = 0.0062 43 F D D 2 PCOD4500048 SiO2 R = 0.0051 43 F D D 2 PCOD4500049 SiO2 R = 0.0048 43 F D D 2 PCOD4500031 SiO2 R = 0.0041 36 C M C 21 2 times the same model PCOD4500029 SiO2 R = 0.0065 36 C M C 21 PCOD4500011 SiO2 R = 0.0045 20 C 2 2 21 3 times the same model PCOD4500008 SiO2 R = 0.0067 20 C 2 2 21 PCOD4500009 SiO2 R = 0.0063 20 C 2 2 21 EtcFinally, CONNECT is performing the same task as GRINSP itself when the list of unique models is identified at the end of the .imp file.
=============================================================================== FINAL LIST OF UNIQUE PROPOSALS, sorted by R : =============================================================================== R NT Vol FD a b c alpha beta gamma MC Run File Ident? 0.0013 8 361.02 22.1597 6.9675 6.9710 7.4328 90.000 90.000 90.000 2 89 700008 CRISTOBALIT 0.0027 8 368.59 21.7045 8.9135 4.8747 8.4830 90.000 90.000 90.000 7762 1514 440026 PCOD 710001 0.0032 12 450.62 26.6301 4.9265 8.6867 10.5298 90.000 90.000 90.000 153 1782 200048 QUARTZ 0.0033 32 1945.19 16.4508 13.8758 10.1564 13.8026 90.000 90.000 90.000 1501 1406 220103 GIS 0.0034 8 428.02 18.6906 5.0921 10.1408 8.2890 90.000 90.000 90.000 534 1201 520012 ABW 0.0034 12 494.19 24.2820 5.0553 10.8859 8.9802 90.000 90.000 90.000 426 1602 450029 PCOD 740021 0.0034 8 335.84 23.8210 8.0582 5.0521 8.2494 90.000 90.000 90.000 65 385 260004 TRIDYMITE 0.0040 16 596.60 26.8187 8.9009 14.2210 4.7132 90.000 90.000 90.000 52 223 430017 PCOD 700002 0.0041 16 836.90 19.1182 9.5155 9.9582 8.8321 90.000 90.000 90.000 1491 614 360019 PCOD 630014 0.0042 12 648.68 18.4991 8.6590 8.6618 8.6488 90.000 90.000 90.000 3902 1755 230051 SOD 0.0045 12 587.34 20.4310 7.1288 11.7931 6.9862 90.000 90.000 90.000 3624 1082 200023 PCOD 200010 0.0046 16 737.58 21.6924 9.0896 9.5722 8.4773 90.000 90.000 90.000 51 1919 360053 PCOD 640003 0.0047 12 564.22 21.2682 7.3123 15.6050 4.9446 90.000 90.000 90.000 4281 231 630004 BIK 0.0048 16 714.58 22.3909 5.2489 14.9206 9.1242 90.000 90.000 90.000 281 42 690001 PCOD 720011 0.0048 16 695.25 23.0132 15.8401 8.8016 4.9868 90.000 90.000 90.000 2553 1155 400013 PCOD 400005 0.0049 14 707.83 19.7789 6.3038 7.2929 15.3965 90.000 90.000 90.000 3582 429 230010 PCOD 230010 0.0051 12 559.51 21.4472 8.9773 10.3248 6.0365 90.000 90.000 90.000 72 1654 670004 PCOD 670003 0.0055 10 608.49 16.4342 9.7613 9.7443 6.3973 90.000 90.000 90.000 85191 993 180002 EDI 0.0056 8 319.50 25.0391 5.2417 4.8038 12.6887 90.000 90.000 90.000 28 1246 240044 PCOD 240044 0.0058 40 1870.96 21.3793 10.3170 13.1406 13.8006 90.000 90.000 90.000 726 712 730001 PCOD 730001 0.0058 10 376.79 26.5401 6.4514 4.6984 12.4307 90.000 90.000 90.000 7235 521 230014 PCOD 230014 0.0059 20 1227.67 16.2910 13.7750 13.8734 6.4240 90.000 90.000 90.000 50834 744 240029 NAT 0.0059 12 580.57 20.6694 13.0594 9.1383 4.8648 90.000 90.000 90.000 2122 62 200004 PCOD 200004 0.0060 8 306.70 26.0838 8.9958 7.2965 4.6726 90.000 90.000 90.000 28 1553 200042 PCOD 200002 0.0061 20 915.21 21.8530 14.3208 5.1496 12.4102 90.000 90.000 90.000 20941 1438 670002 PCOD 670002 0.0061 16 758.36 21.0981 8.7498 16.8682 5.1382 90.000 90.000 90.000 100 1557 360046 PCOD 360031 0.0063 20 1090.25 18.3444 9.5394 16.5853 6.8910 90.000 90.000 90.000 127096 1732 550007 PCOD 550007 0.0064 10 429.96 23.2580 8.6555 4.5462 10.9267 90.000 90.000 90.000 2032 1259 230038 PCOD 230002 0.0067 24 1211.09 19.8169 16.0871 5.2582 14.3174 90.000 90.000 90.000 3848 305 690003 PCOD 690003 0.0067 12 597.91 20.0698 7.5333 5.0628 15.6770 90.000 90.000 90.000 179 127 570002 JBW 0.0068 16 933.34 17.1428 10.1442 9.3739 9.8153 90.000 90.000 90.000 36415 1751 440032 ACO 0.0069 24 1144.78 20.9646 11.4090 11.1425 9.0052 90.000 90.000 90.000 4846 1317 370006 PCOD 370003 0.0070 8 326.70 24.4872 8.5680 8.1240 4.6935 90.000 90.000 90.000 715 1090 330027 PCOD 330014 0.0070 28 1544.08 18.1338 8.2739 15.1687 12.3030 90.000 90.000 90.000 4318 1755 720013 PCOD 720013 0.0071 6 323.33 18.5567 7.8229 5.2452 7.8800 90.000 90.000 90.000 44943 301 250001 PCOD 250001 0.0072 32 1588.64 20.1430 8.1375 13.9744 13.9703 90.000 90.000 90.000 1929 197 360008 PCOD 450025 0.0073 12 622.96 19.2629 5.1841 11.1652 10.7627 90.000 90.000 90.000 1733 818 190026 PCOD 620004 0.0074 20 807.61 24.7643 6.7322 13.1351 9.1330 90.000 90.000 90.000 1616 689 450013 PCOD 450013 0.0075 24 1168.17 20.5449 14.9664 8.7792 8.8907 90.000 90.000 90.000 18663 1669 440029 PCOD 460031 0.0076 16 734.95 21.7702 8.6236 5.0654 16.8249 90.000 90.000 90.000 17121 453 310008 PCOD 310008 0.0076 10 342.44 29.2024 12.3922 6.1469 4.4955 90.000 90.000 90.000 2 199 230005 PCOD 230005 0.0076 32 1756.93 18.2136 14.3148 14.3022 8.5816 90.000 90.000 90.000 5657 1296 450026 PCOD 450026 0.0081 12 521.25 23.0216 10.9829 9.1328 5.1967 90.000 90.000 90.000 80 803 390002 PCOD 390002 0.0082 9 501.64 17.9413 7.9015 7.8104 8.1285 90.000 90.000 90.000 147639 1351 250003 PCOD 250003 0.0083 16 824.98 19.3943 9.2286 13.2011 6.7717 90.000 90.000 90.000 165260 413 190017 PCOD 190017 0.0083 12 468.51 25.6133 6.6836 10.0733 6.9588 90.000 90.000 90.000 5405 1482 600010 PCOD 600010 0.0084 20 1169.31 17.1041 5.1393 12.5137 18.1818 90.000 90.000 90.000 177422 1479 190057 PCOD 190057 0.0085 16 789.34 20.2700 12.4578 12.0723 5.2485 90.000 90.000 90.000 49 178 400003 PCOD 400003 0.0085 16 829.08 19.2985 9.5249 6.6388 13.1115 90.000 90.000 90.000 8163 499 290005 PCOD 290005 0.0086 16 818.66 19.5442 8.9983 17.2871 5.2628 90.000 90.000 90.000 2228 659 360022 PCOD 360022 0.0087 16 663.31 24.1214 12.7331 4.8813 10.6720 90.000 90.000 90.000 109 415 460018 PCOD 460018 0.0090 20 916.61 21.8196 13.6697 6.6169 10.1337 90.000 90.000 90.000 1796 1276 560003 PCOD 560003 0.0090 12 498.71 24.0621 7.5668 13.2404 4.9778 90.000 90.000 90.000 9810 526 190019 PCOD 190019 0.0090 16 641.74 24.9322 11.5305 5.0206 11.0856 90.000 90.000 90.000 19313 974 260007 PCOD 260007 0.0092 12 491.74 24.4030 5.1404 9.8008 9.7606 90.000 90.000 90.000 3225 201 190010 PCOD 190010 0.0092 8 570.95 14.0117 13.1227 5.3287 8.1650 90.000 90.000 90.000 77244 1411 390005 PCOD 390005 0.0093 20 976.55 20.4802 11.7140 12.3496 6.7505 90.000 90.000 90.000 61708 1914 560004 PCOD 560004 0.0094 16 664.25 24.0874 8.8854 5.9774 12.5067 90.000 90.000 90.000 8482 1448 290016 PCOD 290016 0.0096 12 501.60 23.9236 5.2949 6.8507 13.8281 90.000 90.000 90.000 6 167 540001 PCOD 730002 0.0097 32 1634.85 19.5737 10.4747 9.2333 16.9035 90.000 90.000 90.000 6312 1585 700162 MON 14-Apr-2006 3 hour 9 min 26 Sec Total CPU time elapsed in seconds : 31422.70 Total number of runs : 118059 Number of structure candidates : 94963 Number of them with Rdt0 < 0.1400000 is : 45625 Number of them with Rdt0 < 0.1300000 is : 33563 Number of them with Rdt0 < 0.1200000 is : 23689 Number of them with Rdt0 < 0.1100000 is : 14837 Number of them with Rdt0 < 0.1000000 is : 7815 Successful optimizations : 1484 Unique proposals : 60The difference between 60 models from GRINSP and the 48 models found by CONNECT is explained by the fact that the calculation with GRINSP avoided to save the CIFs of the models already identified into the file connectivity.txt (the known zeolites and dense SiO2 phases). The list above show that 12 known models were retrieved (Cristobalite, Quartz, GIS, ABW, Trydimite, SOD, BIK, EDI, NAT, JBW, ACO, MON).
CONNECT produces also a new .con file with exclusively the best models.
CONNECT will help you to make a global analysis of a total list of CIF files (the lists studied here was corrsponding to a small time calculation restricted to orthorhombic space groups).
Complex job ?
Yes !...
FRAMDENS
Satellite software allowing to read a multiple CIF and to provide a list (in a file with .out extension) of models ordered according to the framework densities (from the smallest to the largest).
The example inside of framdens.zip is the
same total.cif file as above.
Note that this version is only working
for oxides, not for flourides or anything else (because the way to count
the number of cations is by detecting the first O atom appearing in the
list of atomic coordinates...). Moreover, it will not work also if the
cation is Os... A part of the output file is below. Includes are the formula,
the PCOD number the R factor, the volume and finally the framework density
(FD) :
Models ordered according to FD SiO2 PCOD4500040 R = 0.0092 570.95 14.01 SiO2 PCOD4500004 R = 0.0084 1169.31 17.10 SiO2 PCOD4500066 R = 0.0072 893.65 17.90 SiO2 PCOD4500020 R = 0.0082 501.64 17.94 SiO2 PCOD4500085 R = 0.0070 1544.08 18.13 SiO2 PCOD4500029 R = 0.0065 879.87 18.18 SiO2 PCOD4500055 R = 0.0076 1756.93 18.21 SiO2 PCOD4500061 R = 0.0063 1090.25 18.34 SiO2 PCOD4500019 R = 0.0071 323.33 18.56
Output files
From and input file name.dat, GRINSP and GRINS create several output files :
name.imp : summarizes all the results
(including FD, the framework density)
name.con : contains the coordination
sequences (CS) of all predicted unique final models
filename.dat : atomic
coordinates in STRUPLO/STRUVIR
format, allowing a direct look
at the structure in VRML
filename.cif : atomic coordinates
in the IUCr
CIF format
name will be the same as the starting
name.dat file
(i.e. if you start with the parameters file SiO2.dat, then you will obtain
SiO2.imp and
SiO2.con)
filename is determined by the space
group number
working with SG N°230, you will obtain filenames starting at 2300001.cif
and .dat, and next.
working with SG N°1, you will obtain filenames starting at 10001.cif
and .dat, and next.
Be carefull not to
destroy previous series of files with same filenames...
Make runs in different
directories.
Strategy, comments
General prediction
If the aim is to explore the possible crystal structures showing corner-sharing
in M2X3 (triangles) , MX2 (tetrahedra,
square plane), M2X5 (square pyramids or triangular
bipyramids) MX3 (octahedra, trigonal prisms, pentagonal pyramids)
or mixte compounds MaM'bXc, then it is
recommended to make a complete search (SG: 1 to 230), going from high symmetry
to low symmetry. The problem is that this will need long calculation times
if you wish to attain some 10.000 different cells testes per space group.
So that you may decompose the problem into parts. For instance making 10
times calculations for 1000 different cells for the 6 symmetries apart
:
Cubic : 195-230
Hexagonal/trigonal/rhombohedral : 143-194
Tetragonal : 75-142
Orthorhombic : 16-74
Monoclinic : 3-15
Triclinic : 1-2
Anyway, this will need close to 230 days, so that you may choose as well to make a space group per day...
Finally you will have to gather all results and perform a global analysis by using the satellite programs (CRYS2CON, CONNECT, FRAMDENS).
Or you may keep all models (several possibilities of cells and space group per structure type), if you wish.
Using GRINSP as a structure determination tool for zeolites or MX3 compounds or others
If the aim is to explore a small range of cell parameters and if the symmetry is known, this is even more simple. Restrain the search to the known cell parameters (allow a +- 1 Angstrom tolerance).
Two-dimensionnal models
GRINSP may generate two-dimensionnal structures as well as the three-dimensionnal ones, in some cases (with triangles and tetrahedra). These structures were not included into the PCOD, with one exception for a tubular B2O3 predicted model. The problem with these two-dimensionnal models is that GRINSP has no way to estimate the correct distance between the layers or the rods.
Parameters having large influence on the speed
of calculations:
These parameters are mainly nmax, the max cell parameters, nruns, genmax,
Rmax, Rdt0 and idls :
Zeolites SG: 127-133 : text for this run 127 133 : SG : Space groups range (two values between 1 230) 1 0 1 192 : npol, ncon, nmim, nmax 4 : ncpol (coordination for each polyhedron-type) Si O : elements for a search in the distgrinsp.txt file 3. 30. 3. 30. 3. 30. : min and max cell parameters a, b, c 5. 35. : min and max framework density (FD) 200 30000 0.02 0.20 : nruns, genmax, Rmax, Rdt0 20000 1 : idls (MC cycles for distance refinement) and iref (1 or 0) -1 : code for selecting models to outputSome times and results from various tests with the file edi.dat, on a Pentium D, 3.2 Ghz :
a- Searching essentially for EDI with small cell range : 6. 8. 6. 8. 5.5 7.5 and nmax=20
nruns genmax Rmax Rdt0 idls total time candidates candidates successful unique in seconds (total) Rdt < Rdt0 optimization models 200 300000 0.02 0.20 20000 252 188 174 45 6 200 300000 0.02 0.15 20000 139 194 108 45 5 200 300000 0.02 0.10 20000 51 188 29 20 5 200 30000 0.02 0.10 20000 48 187 38 21 4 200 30000 0.02 0.10 6000 17 192 35 20 5
b- Searching all solutions in P-4m2 with larger cell range : 4 16 4 16 4 16 and nmax=64
nruns genmax Rmax Rdt0 idls total time candidates candidates successful unique in seconds (total) Rdt < Rdt0 optimization models 20000 30000 0.02 0.10 6000 1101 17355 1672 636 13 20000 300000 0.02 0.10 6000 1246 19132 1723 640 15 20000 300000 0.02 0.12 6000 2711 19093 3567 1031 22 c- with even larger cell range : 4 22 4 22 4 22 and nmax=192
nruns genmax Rmax Rdt0 idls total time candidates candidates successful unique in seconds (total) Rdt < Rdt0 optimization models 20000 300000 0.02 0.12 6000 2570 19396 2294 556 19
Conclusions : - Clearly, changing Rdt0 from 0.20 to 0.10 can save time in the ratio 5 to 1, without changing a lot the number of final unique models (in all cases, the EDI model was retrieved). - Decreasing genmasx from 300000 to 30000 has not a great influence (gain of less than 10% in time). - All this means that the optimization second step is the more consuming time. Discarding those rough models with large Rdt0 save much time, apparently without reducing seriously the number of unique final models, since their optimization is unsuccessful, anyway. However, Rdt=0.12 or 0.13 is probably a better choice than 0.10 which would lead to discard many models, as shown by the second series of tests above, in spite of more than twice a longer time at Rdt0=0.12 than at 0.10.
Results already obtained with GRINSP
Zeolites and dense SiO2
AlPO4
Titanosilicates, titanophosphates,
vanadophosphates,gallophosphates
B2O3
V2O5
MF3
Visit the PCOD, see the What's New page
Bugs and imperfections, comments
Problems with the .dat file for being read by STRUVIR for building VRML files (the limit for one atom-type in STRUVIR being 99, you have to rename those in excess and change their numbers..., quite boring, but the whole prediction stuff is boring...).
Sometimes the formula is not correct in the CIF.
The Coordination Sequence (CS) calculation in GRINSP is not absolutely infallible (but works much better for version 2.00 than for version 1.00)...
Using a Rmax up to 0.05 or 0.10 or larger may make appear some strange structures and even some not so strange (trigonal prisms instead of octahedra, etc).
Polyhedra with 5 vertices can be either square pyramids or triangular bipyramids. The latter cannot correspond to all M-X distances equal, and therefore cannot lead to low R values (requiring all distances equal to the ideal one).
Contact the author if you find bugs or have other 'problems' than the above ones...
The final structure is still described in the P 1 space group, but you will find in the .imp file the general or special positions occupied by the cations in the selected working space group before the final refinement made in the P 1 space group. Apply PLATON there for checking the true symmetry, and CRYSCON for transforming the symmetry from P 1 to the real symmetry.
Have fun with GRINSP !
Copyright © 2003-6 Armel Le Bail