|
Structure Determination by Powder Diffractometry Round Robin - 2 Organized by : A. Le Bail and L.M.D Cranswick
- Results available - - Round Robin closed - |
|
|
Conditions Data Download Step 1 Step 2
|
"Over the past decade, we have moved from the isolated solution
of simple structures to the essentially routine solution of twenty- to
fifty-atom structures... Nowadays, moderately complex pharmaceutical structures
can be solved... Armed with diffraction patterns containing several thousand
reflections, then low-resolution protein structures will be a possibility
and two hundred atom structures may yet become routine."
from : William I.F. David,
A bright future for powder diffraction, Z. Kristallogr. 217 (2002) p. 295 |
| Introduction
Routine solution of 15-30 non-hydrogen atom structures was not demonstrated at the first SDPDRR (Structure determination by Powder Diffractometry Round Robin - see also the CPD Newsletter N°25), in June 1998. If more than 70 potential participants downloaded the data, only two solutions were provided for the pharmaceutical sample (~30 non-H atoms), and none for the inorganic one (15 non-H toms). More than four years later, we observe that the number of structures solved by SDPD remains below 100 per annum, in spite of an explosion of the number of different software available for powder structure solution, commercial as well as in the public domain or even open source (TOPAS, DASH, PowderSolve, ENDEAVOUR, EXPO, MRIA, FOX, ESPOIR, PSSP, OCTOPUS, ROTSEARCH, FOCUS, SAFE, EAGER etc). It is time to verify if SDPD on demand has now really become routine,
meaning that software in other hands than those of their developers can
produce regularly the solutions. Many attendees of the XIXth IUCr Congress,
Geneva (August 2002) were clearly impatient to see this round robin organized.
|
|
| Conditions of the SDPDRR-2
The first SDPDRR was made knowing the cell. Thus, the indexing bottleneck was avoided. It will not be avoided this time, however, the round robin will consist in 2 separated steps : indexing, and structure solution and refinement because experts in indexing could be different from experts in structure solution. At the end of step 1 (indexing), the results will be disclosed so as to allow possibly more participants at step 2. Note however that participants will receive a participant number at step 1, and if they participate too to step 2, they will keep that same participant number. Anonymity will be ensured unless the participants want to disclose their
identity.
|
|
| Data Download
We have prepared 8 samples, all powder patterns are experimental, with the exception of the sample 2 pattern which was calculated from a single crystal structure. Samples 1 to 3 are to be used at steps 1 AND 2 (whole SDPD). Samples 4 to 8 are to be used ONLY at the indexing step 1 (a representative
number of samples showing various problems
Even if not mentioned, impurity presence as well as systematic zeropoint error or even some preferred orientation are possibilities to have in mind... Each sample(n).zip file contains the dataset in various formats (.dat for Winplotr instrum=0, Bruker .raw, Bruker .uxd, GSAS .gsa, XY column .xy, Philips .rd and finlly CPI .cpi formats, plus a .txt file including the peak positions list obtained by using PowderX). Download all 8 samples : samples.zip
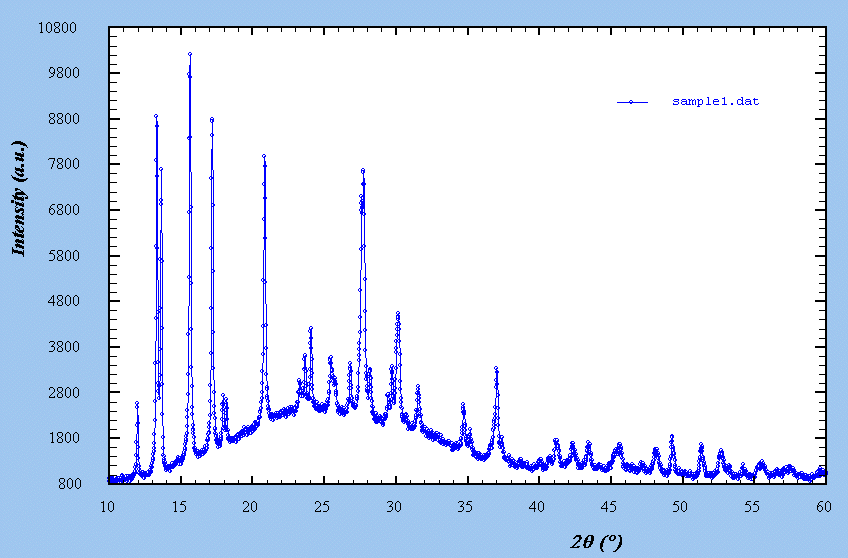
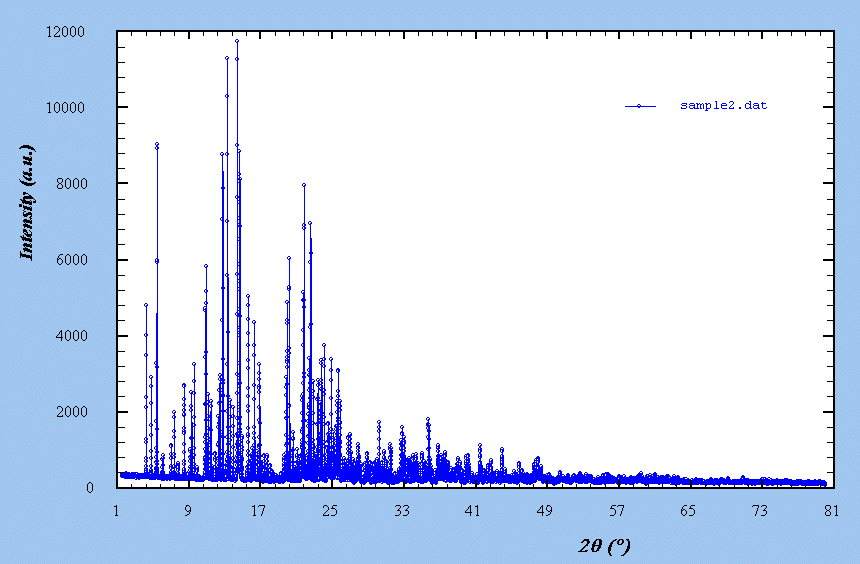
- Probable formula : Al2F10[C6N4H20] - Probable impurity : pyrochlore structure, cubic, Fd3m, a ~ 9.8 A, with possible formula Al(F,OH)3.xH2O - Cartesian coordinates (.xyz file) for the C6N4H20 molecule, as found in C6N4H20.Cl4 - The usual behaviour of Al in F environment is to form AlF6 octahedra - Reflection laboratory X-ray data from a Bruker D8 Advance diffractometer, CuKalpha, very few sample (40 mg) - Data : sample1.zip - See the pattern Sample 2
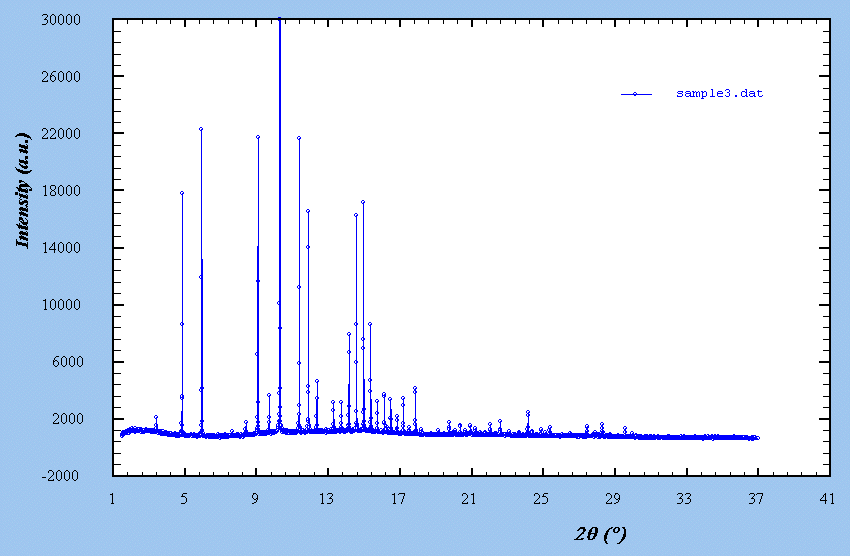
Sample 3
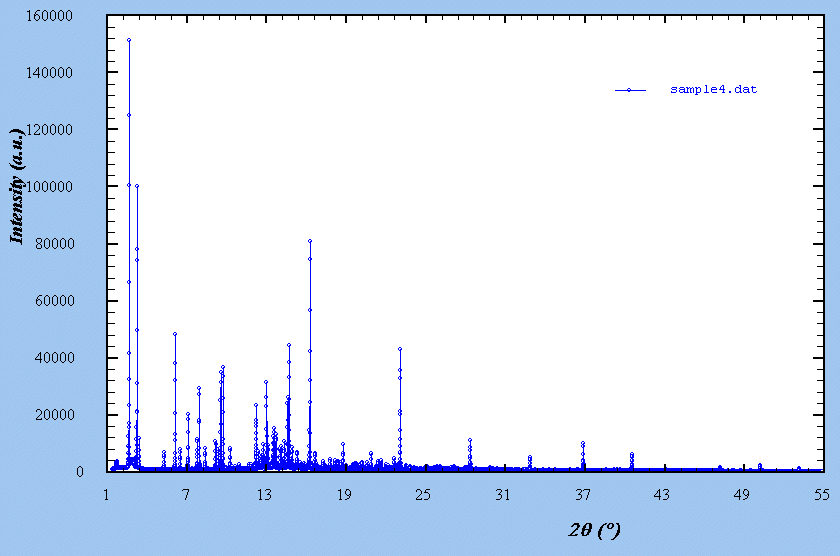
These additional powder patterns are mainly for indexing experts and indexing software developers, for which samples 1-3 would be too easy, and not enough for a serious comparison of indexing software. Sample 4
Sample 5
Sample 6
Sample 7
Sample 8
|
|
| Step 1 - Indexing - CLOSED
Peak position hunting is software- and user-dependent. The round robin participants are encouraged to use the peak hunting software of their choice, though a list of peak positions is furnished for each sample, obtained by using the PowderX software (.txt files), doing our best. We consider that indexing is giving the correct cell parameters, not only a list of possibilities ordered by decreasing value of figures of merit. We expect only one cell proposal from every participants, for each sample (or at least for samples 1-3 from people making SDPDs routinely), together with the list of indexed peaks, and figures of merit, the name of the software (for indexing and peak position hunting), a statement telling if the cell was ascertained by applying CHEKCELL or a whole pattern fitting decomposition program (if yes, the software name and the method name - Pawley or Le Bail fit, etc), and finally a space group proposal (tell if estimated manually or with a software help, give the software name, if any), plus explanations judged necessary (use of databases like CSD, ICSD, ICDD, etc).However, giving a list of possible unchecked cells ordered by decreasing value of figures of merit will be accepted too. All this should be given inside one file (either a PDF or a MS Word document), preferably compressed by Winzip, attached to the email. Be careful to not include your name inside that file if you want to keep anonymity. Robin Shirley passed on CDT files
(converted from the PowderX peak list) ready for using the CRYSFIRE suite
(including 8 indexing software). Have also a look at the new indexing program
McMaille.
Send the results to : L.M.D. Cranswick : l.m.d.cranswick@dl.ac.uk STARTING DATE : Monday 9th September
|
|
| Step 2 - Structure Solution and
Refinement - CLOSED
For samples 1-3 only We expect all details (see and get the form) gathered into a PDF or MS Word document, preferably compressed by Winzip, attached to the email. Be careful to not include your name inside that file if you want to keep anonymity. Send the results to : A. Le Bail : lebail@univ-lemans.fr And after all, if you can also determine the structures of samples 4-8 without the molecular formula of some of them, then, why not ? Routine is supposed to be fast and quasi-automatic. STARTING DATE : Monday 9th September (as soon as you have indexed
samples 1-3 powder patterns -a few minutes-, you may start solving their
structures)
|
|
|
ROUTINE SDPD ? 
But, lost in the maze, which button to press ? And when ? Above is a very partial randomized list of software/methods. A. Le Bail : alb@cristal.org L.M.D. Cranswick : l.m.d.cranswick@dl.ac.uk ************ September-November 2002 Sponsored by the CCP14 and the SDPD Internet Course |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}