Inheritance diagram for ObjCryst::Molecule:
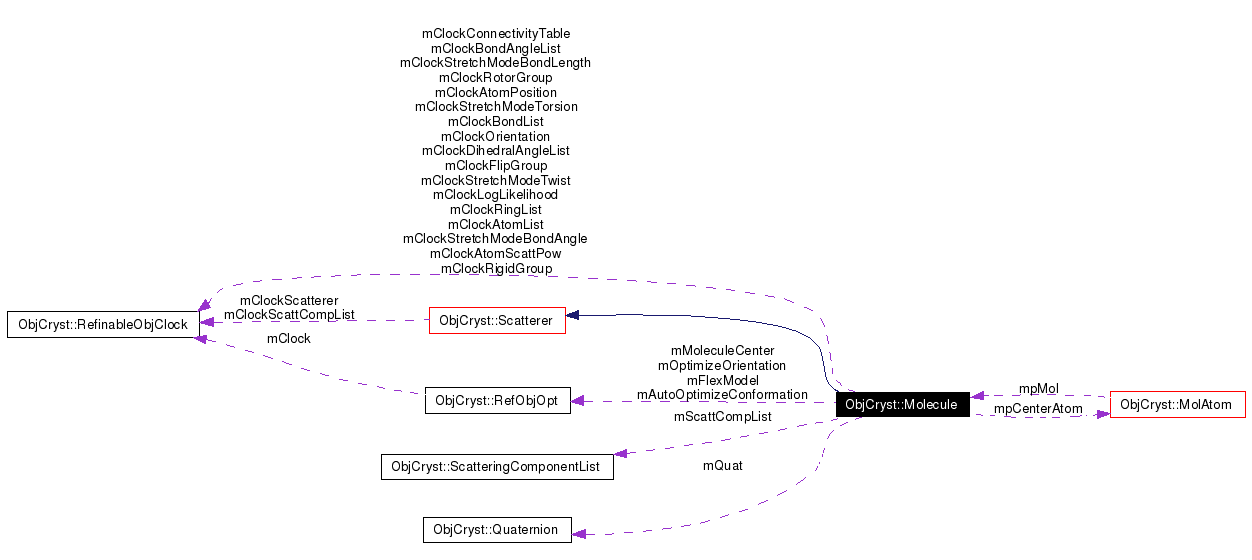
Public Member Functions | |
| Molecule (Crystal &cryst, const string &name="") | |
| Constructor. | |
| Molecule (const Molecule &old) | |
| Copy constructor. | |
| ~Molecule () | |
| Destructor. | |
| virtual Molecule * | CreateCopy () const |
| virtual const string & | GetClassName () const |
| Name for this class ("RefinableObj", "Crystal",...). | |
| virtual void | Print () const |
| Print some info about the scatterer (ideally this should be one line...). | |
| virtual void | XMLOutput (ostream &os, int indent=0) const |
| Output to stream in well-formed XML. | |
| virtual void | XMLInput (istream &is, const XMLCrystTag &tag) |
| Input From stream. | |
| virtual void | BeginOptimization (const bool allowApproximations=false, const bool enableRestraints=false) |
| This should be called by any optimization class at the begining of an optimization. | |
| virtual void | RandomizeConfiguration () |
| Randomize Configuration (before a global optimization). | |
| virtual void | GlobalOptRandomMove (const REAL mutationAmplitude, const RefParType *type) |
| Make a random move of the current configuration. | |
| virtual REAL | GetLogLikelihood () const |
| Get -log(likelihood) of the current configuration for the object. | |
| virtual void | TagNewBestConfig () const |
| During a global optimization, tells the object that the current config is the latest "best" config. | |
| virtual int | GetNbComponent () const |
| Number of components in the scatterer (eg number of point scatterers). | |
| virtual const ScatteringComponentList & | GetScatteringComponentList () const |
| Get the list of all scattering components for this scatterer. | |
| virtual string | GetComponentName (const int i) const |
| Name for the i-th component of this scatterer. | |
| virtual ostream & | POVRayDescription (ostream &os, const CrystalPOVRayOptions &options) const |
| virtual void | GLInitDisplayList (const bool onlyIndependentAtoms=false, const REAL xMin=-.1, const REAL xMax=1.1, const REAL yMin=-.1, const REAL yMax=1.1, const REAL zMin=-.1, const REAL zMax=1.1, const bool displayEnantiomer=false, const bool displayNames=false) const |
| void | AddAtom (const REAL x, const REAL y, const REAL z, const ScatteringPower *pPow, const string &name, const bool updateDisplay=true) |
| Add an atom. | |
| vector< MolAtom * >::iterator | RemoveAtom (MolAtom &) |
| Remove an atom. | |
| void | AddBond (MolAtom &atom1, MolAtom &atom2, const REAL length, const REAL sigma, const REAL delta, const REAL bondOrder=1., const bool updateDisplay=true) |
| Add a bond. | |
| vector< MolBond * >::iterator | RemoveBond (const MolBond &) |
| Remove a bond. | |
| vector< MolBond * >::const_iterator | FindBond (const MolAtom &, const MolAtom &) const |
| Searches whether a bond between two atoms already exists. | |
| vector< MolBond * >::iterator | FindBond (const MolAtom &, const MolAtom &) |
| Searches whether a bond between two atoms already exists. | |
| void | AddBondAngle (MolAtom &atom1, MolAtom &atom2, MolAtom &atom3, const REAL angle, const REAL sigma, const REAL delta, const bool updateDisplay=true) |
| Add a bond angle restraint. | |
| vector< MolBondAngle * >::iterator | RemoveBondAngle (const MolBondAngle &) |
| Remove a BondAngle. | |
| vector< MolBondAngle * >::const_iterator | FindBondAngle (const MolAtom &at1, const MolAtom &at0, const MolAtom &at2) const |
| Searches whether a bond between three atoms already exists, searching for either (at1,at2,at3) and (at3,at2,at1), as these are equivalent. | |
| void | AddDihedralAngle (MolAtom &atom1, MolAtom &atom2, MolAtom &atom3, MolAtom &atom4, const REAL angle, const REAL sigma, const REAL delta, const bool updateDisplay=true) |
| Add a dihedral angle restraint. | |
| vector< MolDihedralAngle * >::iterator | RemoveDihedralAngle (const MolDihedralAngle &) |
| Remove a dihedral angle. | |
| vector< MolDihedralAngle * >::const_iterator | FindDihedralAngle (const MolAtom &at1, const MolAtom &at2, const MolAtom &at3, const MolAtom &at4) const |
| Searches whether a dihedral between four atoms already exists, searching for either (at1,at2,at3,at4) and (at4,at3,at2,at1), as these are equivalent. | |
| void | AddRigidGroup (const RigidGroup &, const bool updateDisplay=true) |
| Add a rigid group of atoms. | |
| std::vector< RigidGroup * >::iterator | RemoveRigidGroup (const RigidGroup &group, const bool updateDisplay=true) |
| Remove a rigid group of atoms. | |
| MolAtom & | GetAtom (unsigned int i) |
| const MolAtom & | GetAtom (unsigned int i) const |
| MolAtom & | GetAtom (const string &name) |
| const MolAtom & | GetAtom (const string &name) const |
| vector< MolAtom * >::reverse_iterator | FindAtom (const string &name) |
| Search a MolAtom from its name. | |
| vector< MolAtom * >::const_reverse_iterator | FindAtom (const string &name) const |
| Search a MolAtom from its name. | |
| void | OptimizeConformation (const long nbTrial=10000, const REAL stopCost=0.) |
| Minimize configuration from internal restraints (bond lengths, angles and dihedral angles). | |
| const vector< MolAtom * > & | GetAtomList () const |
| const vector< MolBond * > & | GetBondList () const |
| const vector< MolBondAngle * > & | GetBondAngleList () const |
| const vector< MolDihedralAngle * > & | GetDihedralAngleList () const |
| vector< MolAtom * > & | GetAtomList () |
| vector< MolBond * > & | GetBondList () |
| vector< MolBondAngle * > & | GetBondAngleList () |
| vector< MolDihedralAngle * > & | GetDihedralAngleList () |
| list< StretchModeBondLength > & | GetStretchModeBondLengthList () |
| list< StretchModeBondAngle > & | GetStretchModeBondAngleList () |
| list< StretchModeTorsion > & | GetStretchModeTorsionList () |
| const list< StretchModeBondLength > & | GetStretchModeBondLengthList () const |
| const list< StretchModeBondAngle > & | GetStretchModeBondAngleList () const |
| const list< StretchModeTorsion > & | GetStretchModeTorsionList () const |
| const std::vector< RigidGroup * > & | GetRigidGroupList () const |
| List of rigid group of atoms. | |
| std::vector< RigidGroup * > & | GetRigidGroupList () |
| List of rigid group of atoms. | |
| void | RotateAtomGroup (const MolAtom &at1, const MolAtom &at2, const set< MolAtom * > &atoms, const REAL angle, const bool keepCenter=true) |
| Rotate a group of atoms around an axis defined by two atoms. | |
| void | RotateAtomGroup (const MolAtom &at, const REAL vx, const REAL vy, const REAL vz, const set< MolAtom * > &atoms, const REAL angle, const bool keepCenter=true) |
| Rotate a group of atoms around an axis defined by one atom and a vector. | |
| void | TranslateAtomGroup (const set< MolAtom * > &atoms, const REAL dx, const REAL dy, const REAL dz, const bool keepCenter=true) |
| Translate a group of atoms in a given direction. | |
| void | RestraintStatus (ostream &os) const |
| Print the status of all restraints (bond length, angles...). | |
| const map< MolAtom *, set< MolAtom * > > & | GetConnectivityTable () |
| Get the connectivity table. | |
| RefinableObjClock & | GetBondListClock () |
| get the clock associated to the list of bonds | |
| const RefinableObjClock & | GetBondListClock () const |
| get the clock associated to the list of bonds | |
| RefinableObjClock & | GetAtomPositionClock () |
| Get the clock associated to the atomic positions. | |
| const RefinableObjClock & | GetAtomPositionClock () const |
| Get the clock associated to the atomic positions. | |
| RefinableObjClock & | GetRigidGroupClock () |
| Get the clock associated to the list of rigid groups (clicked also whenever a rigid group is modified). | |
| const RefinableObjClock & | GetRigidGroupClock () const |
| Get the clock associated to the list of rigid groups (clicked also whenever a rigid group is modified). | |
| void | RigidifyWithDihedralAngles () |
| Add dihedral angles so as to rigidify the Molecule. | |
| REAL | BondLengthRandomChange (const StretchModeBondLength &mode, const REAL amplitude, const bool respectRestraint=true) |
| Stretch a bond, while respecting the Restraint (if any). | |
| REAL | BondAngleRandomChange (const StretchModeBondAngle &mode, const REAL amplitude, const bool respectRestraint=true) |
| change a bond angle, while respecting the Restraint (if any). | |
| REAL | DihedralAngleRandomChange (const StretchModeTorsion &mode, const REAL amplitude, const bool respectRestraint=true) |
| Change a dihedral angle, while respecting the Restraint (if any). | |
| const MolAtom * | GetCenterAtom () const |
| Get the atom defining the origin of the Molecule Equal to 0 if no atom as been set. | |
| void | SetCenterAtom (const MolAtom &at) |
| Get the atom defining the origin of the Molecule Equal to 0 if no atom as been set. | |
| const std::vector< MolZAtom > & | AsZMatrix (const bool keeporder) const |
| Molecule as Z-matrix. | |
| virtual void | InitRefParList () |
| Prepare refinable parameters for the scatterer object. | |
| void | BuildRingList () |
| Build the list of rings in the molecule. | |
| void | BuildConnectivityTable () const |
| Build the Connectivity table. | |
| void | BuildRotorGroup () |
| Build the groups of atoms that will be rotated during global optimization. | |
| void | TuneGlobalOptimRotationAmplitude () |
| Tune the rotation amplitude for free torsions and for the overall Molecule Rotation. | |
| void | BuildFlipGroup () |
| Build the groups of atoms that can be flipped. | |
| void | BuildStretchModeBondLength () |
| Build the groups of atoms moved when stretching a bond length, while respecting the Molecule restraints. | |
| void | BuildStretchModeBondAngle () |
| Build the groups of atoms moved when changing a bond angle, while respecting the Molecule restraints. | |
| void | BuildStretchModeTorsion () |
| Build the groups of atoms moved when changing a dihedral angle, while respecting the Molecule restraints. | |
| void | BuildStretchModeTwist () |
| Build the groups of atoms used to twist internally the Molecule, e.g. | |
| void | BuildStretchModeGroups () |
| Separate StretchMode that break more than their assigned restraint from others. | |
| void | UpdateScattCompList () const |
| Update the Molecule::mScattCompList from the cartesian coordinates of all atoms, and the orientation parameters. | |
| void | InitOptions () |
| Build options for this object. | |
| void | FlipAtomGroup (const FlipGroup &) |
| Flip a group of atom. See Molecule::FlipGroup. | |
Public Attributes | |
| ScatteringComponentList | mScattCompList |
| The list of scattering components. | |
| vector< MolAtom * > | mvpAtom |
| The list of atoms. | |
| vector< MolBond * > | mvpBond |
| The list of bonds. | |
| vector< MolBondAngle * > | mvpBondAngle |
| The list of bond angles. | |
| vector< MolDihedralAngle * > | mvpDihedralAngle |
| The list of dihedral angles. | |
| map< MolAtom *, std::vector< MolBond * > > | mvAtomBond |
| List of Bonds for each atom. | |
| std::vector< RigidGroup * > | mvRigidGroup |
| Rigid groups of atoms. | |
| list< MolRing > | mvRing |
| The list of rings. | |
| Quaternion | mQuat |
| The unit quaternion defining the orientation. | |
| REAL | mBaseRotationAmplitude |
| Base Rotation amplitude (in radians) for the Molecule, so that the average atomic displacement is equal to 0.1 A. | |
| RefinableObjClock | mClockAtomList |
| RefinableObjClock | mClockBondList |
| RefinableObjClock | mClockBondAngleList |
| RefinableObjClock | mClockDihedralAngleList |
| RefinableObjClock | mClockRigidGroup |
| RefinableObjClock | mClockAtomPosition |
| RefinableObjClock | mClockAtomScattPow |
| RefinableObjClock | mClockOrientation |
| RefinableObjClock | mClockLogLikelihood |
| RefinableObjClock | mClockConnectivityTable |
| RefinableObjClock | mClockRingList |
| RefinableObjClock | mClockRotorGroup |
| RefinableObjClock | mClockFlipGroup |
| RefinableObjClock | mClockStretchModeBondLength |
| RefinableObjClock | mClockStretchModeBondAngle |
| RefinableObjClock | mClockStretchModeTorsion |
| RefinableObjClock | mClockStretchModeTwist |
| unsigned long | mLocalParamSet |
| unsigned long | mRandomConformChangeNbTest |
| unsigned long | mRandomConformChangeNbAccept |
| REAL | mRandomConformChangeTemp |
| REAL | mLastLogLike |
| bool | mIsSelfOptimizing |
| RefObjOpt | mFlexModel |
| OPtion for the different types of flexibility possible for this molecule: rigid body, free atoms + restraints, torsion angles... | |
| RefObjOpt | mAutoOptimizeConformation |
| Option to automatically optimize the starting conformation, if the total restraint cost is too high. | |
| RefObjOpt | mOptimizeOrientation |
| Option to optimize the Molecule's orientation. | |
| RefObjOpt | mMoleculeCenter |
| Option to choose the center of rotation of the Molecule for the global orientation either as the geometrical center, or as a given atom. | |
| const MolAtom * | mpCenterAtom |
| Atom chosen as center of rotation, if mRotationCenter is set to use an atom rather than the geometrical center. | |
| map< MolAtom *, set< MolAtom * > > | mConnectivityTable |
| Connectivity table: for each atom, keep the list of atoms bonded to it. | |
| list< RotorGroup > | mvRotorGroupTorsion |
| List of RotorGroups corresponding to free torsion bonds. | |
| list< RotorGroup > | mvRotorGroupTorsionSingleChain |
| List of RotorGroups corresponding to free torsion bonds, but with only one chain of atoms listed. | |
| list< RotorGroup > | mvRotorGroupInternal |
| List of RotorGroups for internal rotations. | |
| list< FlipGroup > | mvFlipGroup |
| The list of FlipGroups. | |
| list< StretchModeBondLength > | mvStretchModeBondLength |
| List of StretchModeBondLength. | |
| list< StretchModeBondAngle > | mvStretchModeBondAngle |
| List of StretchModeBondLength. | |
| list< StretchModeTorsion > | mvStretchModeTorsion |
| List of StretchModeBondLength. | |
| list< StretchModeTwist > | mvStretchModeTwist |
| List of StretchModeTwist. | |
| std::list< StretchMode * > | mvpStretchModeFree |
| Groups of StretchMode not breaking any restraint (unless the one they are associated to). | |
| std::list< StretchMode * > | mvpStretchModeNotFree |
| Groups of StretchMode breaking restraints (beyond the one they are associated to). | |
| std::vector< MolZAtom > | mAsZMatrix |
| The Molecule, as a lightweight ZMatrix, for export purposes. | |
| REAL | mLogLikelihood |
| The current log(likelihood). | |
This can also be used for non-organic compounds (polyhedras etc...)
: all atoms must be somehow connected
|
||||||||||||
|
Constructor.
|
|
|
Copy constructor.
|
|
|
Destructor.
|
|
||||||||||||||||||||||||||||
|
Add an atom.
|
|
||||||||||||||||||||||||||||||||
|
Add a bond.
|
|
||||||||||||||||||||||||||||||||
|
Add a bond angle restraint.
|
|
||||||||||||||||||||||||||||||||||||
|
Add a dihedral angle restraint.
|
|
||||||||||||
|
Add a rigid group of atoms. |
|
|
Molecule as Z-matrix.
|
|
||||||||||||
|
This should be called by any optimization class at the begining of an optimization. This will also check that everything is ready, eg call the RefinableObj::Prepare() function. This also affects all sub-objects.
Reimplemented from ObjCryst::RefinableObj. |
|
||||||||||||||||
|
change a bond angle, while respecting the Restraint (if any).
|
|
||||||||||||||||
|
Stretch a bond, while respecting the Restraint (if any).
|
|
|
Build the Connectivity table.
|
|
|
Build the groups of atoms that can be flipped. This is not const because we temporarily modify the molecule conformation to test which FlipGroups are forbidden by restraints (but it should be const). |
|
|
Build the list of rings in the molecule. The list is only rebuilt if the bond or atom list has changed,so it should be safe to call again this function.
|
|
|
Build the groups of atoms that will be rotated during global optimization. This is not const because we temporarily modify the molecule conformation to test which RotorGroups are forbidden by restraints (but it should be const). |
|
|
Build the groups of atoms moved when changing a bond angle, while respecting the Molecule restraints.
|
|
|
Build the groups of atoms moved when stretching a bond length, while respecting the Molecule restraints.
|
|
|
Separate StretchMode that break more than their assigned restraint from others. See Molecule::mvpStretchModeFree and Molecule::mvpStretchModeNotFree |
|
|
Build the groups of atoms moved when changing a dihedral angle, while respecting the Molecule restraints.
|
|
|
Build the groups of atoms used to twist internally the Molecule, e.g. by rotating one chain of atoms between 2 given atoms. |
|
|
For internal use only. so-called Virtual copy constructor, needed to make copies of arrays of Scatterers Implements ObjCryst::Scatterer. |
|
||||||||||||||||
|
Change a dihedral angle, while respecting the Restraint (if any).
|
|
|
Search a MolAtom from its name. Search begins at the end, and the first match is returned. returns mvAtom.rend() if no atom matches |
|
|
Search a MolAtom from its name. Search begins at the end, and the first match is returned. returns mvAtom.rend() if no atom matches |
|
||||||||||||
|
Searches whether a bond between two atoms already exists. If no bond is found, returns Molecule::mvpAtom.end(). |
|
||||||||||||
|
Searches whether a bond between two atoms already exists. If no bond is found, returns Molecule::mvpAtom.end(). |
|
||||||||||||||||
|
Searches whether a bond between three atoms already exists, searching for either (at1,at2,at3) and (at3,at2,at1), as these are equivalent. If no bond angle is found, returns Molecule::mvpBondAngle.end(). |
|
||||||||||||||||||||
|
Searches whether a dihedral between four atoms already exists, searching for either (at1,at2,at3,at4) and (at4,at3,at2,at1), as these are equivalent. If no dihedral angle is found, returns Molecule::mvpDihedralAngle.end(). |
|
|
Flip a group of atom. See Molecule::FlipGroup.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Get the clock associated to the atomic positions.
|
|
|
Get the clock associated to the atomic positions.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
get the clock associated to the list of bonds
|
|
|
get the clock associated to the list of bonds
|
|
|
Get the atom defining the origin of the Molecule Equal to 0 if no atom as been set.
|
|
|
Name for this class ("RefinableObj", "Crystal",...). This is only useful to distinguish different classes when picking up objects from the RefinableObj Global Registry Reimplemented from ObjCryst::Scatterer. |
|
|
Name for the i-th component of this scatterer. If the component is an Atom, Then the name is that of the atom. Else, it is the name of the scatterer plus the component number in the scatterer plus the name of the ScatteringPower.
Implements ObjCryst::Scatterer. |
|
|
Get the connectivity table.
|
|
|
|
|
|
|
|
|
Get -log(likelihood) of the current configuration for the object. By default (no likelihood evaluation available), this is equal to 0. This call should not be recursive, it is the task of the algorithm to get the sum of likelihoods for all objects invlolved.
Reimplemented from ObjCryst::RefinableObj. |
|
|
Number of components in the scatterer (eg number of point scatterers).
Implements ObjCryst::Scatterer. |
|
|
Get the clock associated to the list of rigid groups (clicked also whenever a rigid group is modified).
|
|
|
Get the clock associated to the list of rigid groups (clicked also whenever a rigid group is modified).
|
|
|
List of rigid group of atoms. |
|
|
List of rigid group of atoms. |
|
|
Get the list of all scattering components for this scatterer. This is the most important function of this class, giving the list of scattering positions along with the associated ScatteringPower. Implements ObjCryst::Scatterer. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||||||||||||||
|
For internal use only. Create an OpenGL Display List of the scatterer. This should only be called by a Crystal object.
Implements ObjCryst::Scatterer. |
|
||||||||||||
|
Make a random move of the current configuration. This is for global optimization algorithms. the moves for each parameter are less than their global optimization step, multiplied by the mutation amplitude.
Reimplemented from ObjCryst::RefinableObj. |
|
|
Build options for this object.
|
|
|
Prepare refinable parameters for the scatterer object.
Implements ObjCryst::Scatterer. |
|
||||||||||||
|
Minimize configuration from internal restraints (bond lengths, angles and dihedral angles). Useful when adding manually atoms to get an initial reasonable configuration. |
|
||||||||||||
|
For internal use only. Output a description of the scatterer for POVRay. This should only be called by the Crystal Object to which belongs this scatterer. Implements ObjCryst::Scatterer. |
|
|
Print some info about the scatterer (ideally this should be one line...).
Implements ObjCryst::Scatterer. |
|
|
Randomize Configuration (before a global optimization). This Affects only parameters which are limited and not fixed. The randomization also affects all sub-objects (recursive). Reimplemented from ObjCryst::RefinableObj. |
|
|
Remove an atom. Returns the iterator to the next atom in the list. This also removes all corresponding bonds, bond angles, etc... |
|
|
Remove a bond. Returns the iterator to the next bond in the list. |
|
|
Remove a BondAngle.
|
|
|
Remove a dihedral angle.
|
|
||||||||||||
|
Remove a rigid group of atoms. |
|
|
Print the status of all restraints (bond length, angles...).
|
|
|
Add dihedral angles so as to rigidify the Molecule. In practice, for every sequence of atoms A-B-C-D, add the dihedral angle defined by these 4 atoms, unless either ABC or BCD are aligned (angle below 10°). No duplicate dihedral angle is generated. |
|
||||||||||||||||||||||||||||||||
|
Rotate a group of atoms around an axis defined by one atom and a vector.
|
|
||||||||||||||||||||||||
|
Rotate a group of atoms around an axis defined by two atoms.
|
|
|
Get the atom defining the origin of the Molecule Equal to 0 if no atom as been set.
|
|
|
During a global optimization, tells the object that the current config is the latest "best" config. This can be used by the object to make more intellingent random moves (use with caution: highly experimental !). Reimplemented from ObjCryst::RefinableObj. |
|
||||||||||||||||||||||||
|
Translate a group of atoms in a given direction.
|
|
|
Tune the rotation amplitude for free torsions and for the overall Molecule Rotation. This should be done after Molecule::BuildRotorGroup(); |
|
|
Update the Molecule::mScattCompList from the cartesian coordinates of all atoms, and the orientation parameters.
|
|
||||||||||||
|
Input From stream.
Reimplemented from ObjCryst::RefinableObj. |
|
||||||||||||
|
Output to stream in well-formed XML.
Reimplemented from ObjCryst::RefinableObj. |
|
|
The Molecule, as a lightweight ZMatrix, for export purposes.
|
|
|
Option to automatically optimize the starting conformation, if the total restraint cost is too high. This is done in BeginOptimization(). This is enabled by default, and should be disabled by people who already supply a good starting conformation for their molecule. |
|
|
Base Rotation amplitude (in radians) for the Molecule, so that the average atomic displacement is equal to 0.1 A. Default=0.02*pi |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Connectivity table: for each atom, keep the list of atoms bonded to it. All atoms are referenced from their index. |
|
|
OPtion for the different types of flexibility possible for this molecule: rigid body, free atoms + restraints, torsion angles...
|
|
|
|
|
|
|
|
|
|
|
|
The current log(likelihood).
|
|
|
Option to choose the center of rotation of the Molecule for the global orientation either as the geometrical center, or as a given atom.
|
|
|
Option to optimize the Molecule's orientation. Useful to completely fix the Molecule. |
|
|
Atom chosen as center of rotation, if mRotationCenter is set to use an atom rather than the geometrical center.
|
|
|
The unit quaternion defining the orientation.
|
|
|
|
|
|
|
|
|
|
|
|
The list of scattering components. this is mutable since it only reflects the list of atoms. |
|
|
List of Bonds for each atom. This duplicates the information in Molecule::mvBond |
|
|
The list of FlipGroups.
|
|
|
The list of atoms.
|
|
|
The list of bonds.
|
|
|
The list of bond angles.
|
|
|
The list of dihedral angles.
|
|
|
Groups of StretchMode not breaking any restraint (unless the one they are associated to).
|
|
|
Groups of StretchMode breaking restraints (beyond the one they are associated to).
|
|
|
Rigid groups of atoms. This group will be kept strictly rigid, preventing the use of any stretch mode altering their relative position. The entire group of atoms can however be rotated or translated. |
|
|
The list of rings.
|
|
|
List of RotorGroups for internal rotations. This lists groups of atoms that can be rotated between two given atoms. This is useful to alter the conformation of large rings, where no free torsion bonds exists, and also for long flexible chains. |
|
|
List of RotorGroups corresponding to free torsion bonds. In this list are list of atoms on one side of a bond, that can be rotated freely around this bond. Each bond is listed only once, with the side which has the smallest number of atoms. |
|
|
List of RotorGroups corresponding to free torsion bonds, but with only one chain of atoms listed. The difference with Molecule::mRotorGroupTorsion is that if the bond is A-B, with atom A linked with atoms A1,A2,A3, in this list only one chain (starting either from A1, A2 or A3) will be rotated, instead of the 3 chains. This is useful when searching for the absolute configuration of atoms. |
|
|
List of StretchModeBondLength.
|
|
|
List of StretchModeBondLength.
|
|
|
List of StretchModeBondLength.
|
|
|
List of StretchModeTwist.
|
 1.3.6
1.3.6