Charge Balance restraints on a trivial example
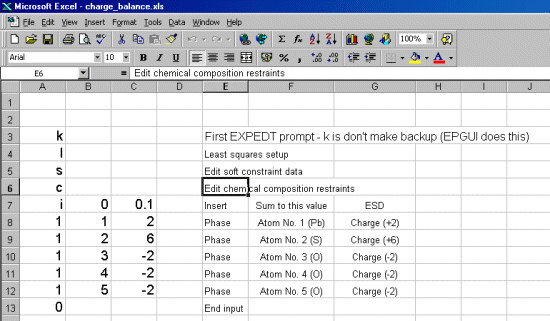
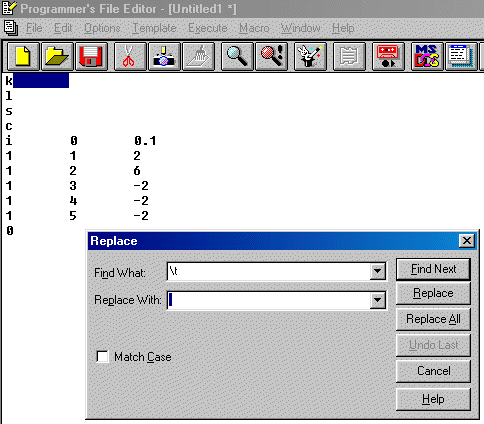
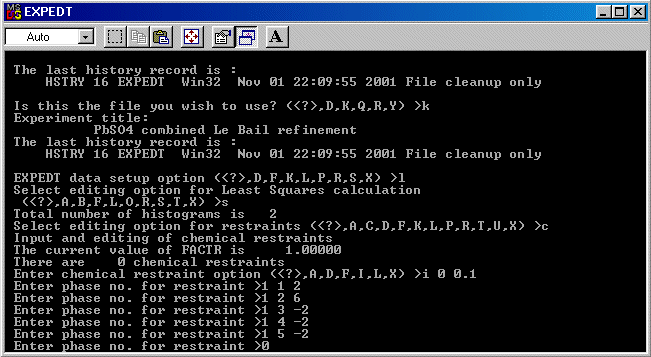
Following is a trivial example involving Lead Sulphate to show how this is done. Obviously, more complex examples with mixed sites could be performed (e.g., Ba+2 vs Fe+3; Ca+2 vs Nb+5).
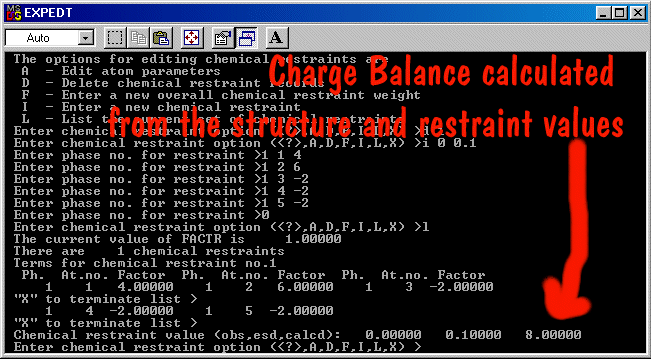
A more complicated example would be that you could define two Fe atoms
on a single site, one (Fe1) defined as Fe+3 and the other (Fe2) as Fe+4. While no
scattering factor for Fe+4 exists, you define the ionization states in the restraints menu
as shown below.
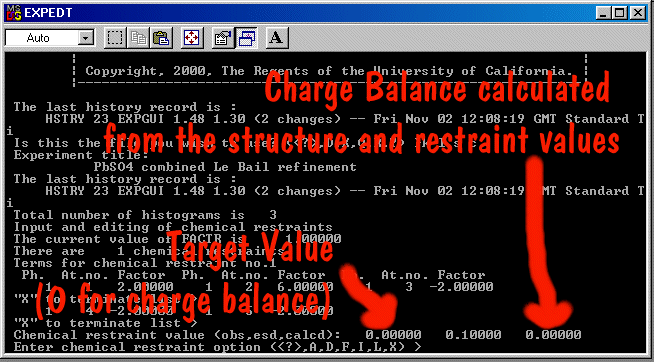
If on multiple sites, combined with the chemistry information (or total chemistry restraints),
you could then refine the amount of Fe+3 vs Fe+4 to achieve charge balance for a particular
structure. (providing you have enough information in the restraints and powder diffraction pattern
to refine the number of unknowns)
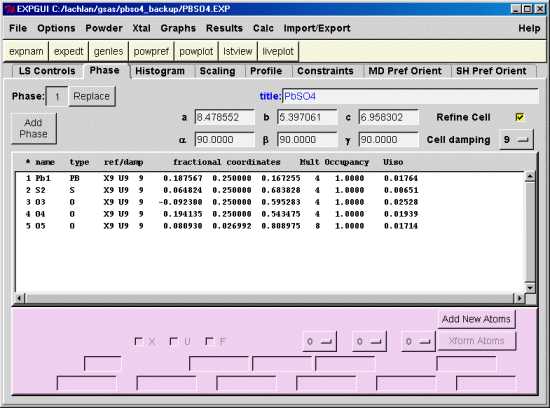
In this case, using Brian Toby's EXPGUI interface into GSAS, note down the atoms numbers and the ionisation states that you will need to assign in the charge balance restraints macro.