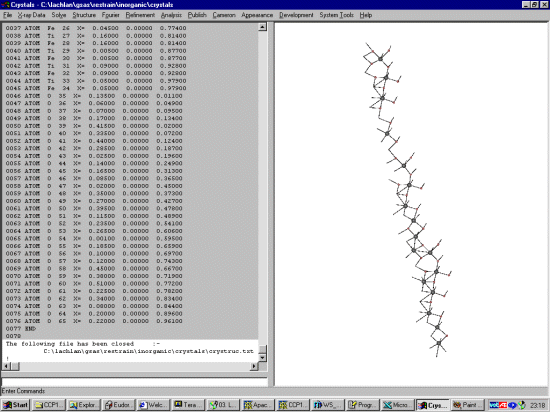
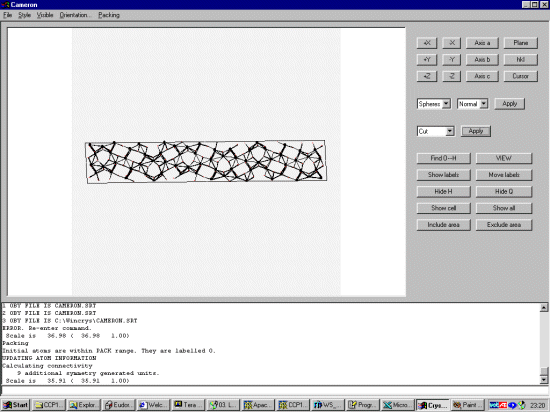
Resulting Crystals Bond Lengths files
You can now globally replace or tweak the bond lengths before
conversion into GSAS Script format using
Scott Belmonte's GPL'd Coue program
for converting Crystals bond-length restraints lists into GSAS macro format. Check the result to
make sure they do match up.
Be wary that you may have to play with the "R" - Set the distance search range as part of the bond-length
restraints menu in GSAS. In this case, setting r=1.5 will get GSAS finding all the bonds correctly.
Make sure you check the results and that everything is working as required
Crystals Format
Again: Be wary that the some Crystals options will not find all the bonds so you have to be wary of this:
e.g., SELECT TYPE=INTRA RANGE=LIMITS may only find what Crystals considers to be
the intramolecular bonds optimised for a discreet molecule - despite this is an extended polymeric framework.
#
# Punched on 10/30/00 at 23:42:46
#
#LIST 16
DIST 2.018,.01= TI(1) to O(35)
DIST 1.822,.01= TI(1) to O(36)
DIST 2.222,.01= TI(1) to O(39)
DIST 2.070,.01= TI(1) to O(40)
DIST 1.929,.01= TI(1) to O(65,-1,2,0,-1,1)
DIST 1.929,.01= TI(1) to O(65,-1,2,0,0,1)
DIST 2.018,.01= FE(2) to O(35)
DIST 1.822,.01= FE(2) to O(36)
DIST 2.222,.01= FE(2) to O(39)
DIST 2.070,.01= FE(2) to O(40)
DIST 1.929,.01= FE(2) to O(65,-1,2,0,-1,1)
DIST 1.929,.01= FE(2) to O(65,-1,2,0,0,1)
DIST 1.900,.01= TI(3) to O(37)
DIST 2.007,.01= TI(3) to O(38)
DIST 2.095,.01= TI(3) to O(40)
DIST 2.159,.01= TI(3) to O(41)
DIST 1.925,.01= TI(3) to O(64,-1,2,0,-1,1)
DIST 1.925,.01= TI(3) to O(64,-1,2,0,0,1)
DIST 1.900,.01= FE(4) to O(37)
DIST 2.007,.01= FE(4) to O(38)
DIST 2.095,.01= FE(4) to O(40)
DIST 2.159,.01= FE(4) to O(41)
DIST 1.925,.01= FE(4) to O(64,-1,2,0,-1,1)
DIST 1.925,.01= FE(4) to O(64,-1,2,0,0,1)
DIST 2.038,.01= TI(5) to O(38)
DIST 2.150,.01= TI(5) to O(42)
DIST 1.985,.01= TI(5) to O(43)
DIST 1.950,.01= TI(5) to O(62,-1,2,0,-1,1)
DIST 1.950,.01= TI(5) to O(62,-1,2,0,0,1)
DIST 1.915,.01= TI(5) to O(63,-1,1,0,0,1)
DIST 2.038,.01= FE(6) to O(38)
DIST 2.150,.01= FE(6) to O(42)
DIST 1.985,.01= FE(6) to O(43)
DIST 1.950,.01= FE(6) to O(62,-1,2,0,-1,1)
DIST 1.950,.01= FE(6) to O(62,-1,2,0,0,1)
DIST 1.915,.01= FE(6) to O(63,-1,1,0,0,1)
DIST 2.209,.01= TI(7) to O(44)
DIST 2.270,.01= TI(7) to O(45)
DIST 1.911,.01= TI(7) to O(56,-1,1,0,0,1)
DIST 2.068,.01= TI(7) to O(57,-1,1,0,0,1)
DIST 2.054,.01= TI(7) to O(59,-1,2,0,-1,1)
DIST 2.054,.01= TI(7) to O(59,-1,2,0,0,1)
DIST 2.209,.01= FE(8) to O(44)
DIST 2.270,.01= FE(8) to O(45)
DIST 1.911,.01= FE(8) to O(56,-1,1,0,0,1)
DIST 2.068,.01= FE(8) to O(57,-1,1,0,0,1)
DIST 2.054,.01= FE(8) to O(59,-1,2,0,-1,1)
DIST 2.054,.01= FE(8) to O(59,-1,2,0,0,1)
DIST 1.991,.01= TI(9) to O(46)
DIST 2.061,.01= TI(9) to O(48)
DIST 2.099,.01= TI(9) to O(49)
DIST 1.925,.01= TI(9) to O(53,-1,2,0,-1,1)
DIST 1.925,.01= TI(9) to O(53,-1,2,0,0,1)
DIST 2.032,.01= TI(9) to O(54,-1,1,0,0,1)
DIST 1.991,.01= FE(10) to O(46)
DIST 2.061,.01= FE(10) to O(48)
DIST 2.099,.01= FE(10) to O(49)
DIST 1.925,.01= FE(10) to O(53,-1,2,0,-1,1)
DIST 1.925,.01= FE(10) to O(53,-1,2,0,0,1)
DIST 2.032,.01= FE(10) to O(54,-1,1,0,0,1)
DIST 1.900,.01= TI(11) to O(47)
DIST 2.038,.01= TI(11) to O(49)
DIST 2.171,.01= TI(11) to O(50)
DIST 2.030,.01= TI(11) to O(51)
DIST 1.965,.01= TI(11) to O(52,-1,2,0,-1,1)
DIST 1.965,.01= TI(11) to O(52,-1,2,0,0,1)
DIST 1.900,.01= FE(12) to O(47)
DIST 2.038,.01= FE(12) to O(49)
DIST 2.171,.01= FE(12) to O(50)
DIST 2.030,.01= FE(12) to O(51)
DIST 1.965,.01= FE(12) to O(52,-1,2,0,-1,1)
DIST 1.965,.01= FE(12) to O(52,-1,2,0,0,1)
DIST 1.833,.01= TI(13) to O(47,-1,1,0,0,1)
DIST 1.950,.01= TI(13) to O(50,-1,2,0,-1,1)
DIST 1.950,.01= TI(13) to O(50,-1,2,0,0,1)
DIST 2.095,.01= TI(13) to O(51)
DIST 1.747,.01= TI(13) to O(51,-1,1,0,0,1)
DIST 2.128,.01= TI(13) to O(52)
DIST 1.833,.01= FE(14) to O(47,-1,1,0,0,1)
DIST 1.950,.01= FE(14) to O(50,-1,2,0,-1,1)
DIST 1.950,.01= FE(14) to O(50,-1,2,0,0,1)
DIST 2.095,.01= FE(14) to O(51)
DIST 1.747,.01= FE(14) to O(51,-1,1,0,0,1)
DIST 2.128,.01= FE(14) to O(52)
DIST 2.033,.01= TI(15) to O(47,-1,1,0,0,1)
DIST 2.122,.01= TI(15) to O(49,-1,2,0,-1,1)
DIST 2.122,.01= TI(15) to O(49,-1,2,0,0,1)
DIST 2.262,.01= TI(15) to O(52)
DIST 2.297,.01= TI(15) to O(53)
DIST 1.825,.01= TI(15) to O(54)
DIST 2.033,.01= FE(16) to O(47,-1,1,0,0,1)
DIST 2.122,.01= FE(16) to O(49,-1,2,0,-1,1)
DIST 2.122,.01= FE(16) to O(49,-1,2,0,0,1)
DIST 2.262,.01= FE(16) to O(52)
DIST 2.297,.01= FE(16) to O(53)
DIST 1.825,.01= FE(16) to O(54)
DIST 1.888,.01= TI(17) to O(46,-1,1,0,0,1)
DIST 1.951,.01= TI(17) to O(48,-1,2,0,-1,1)
DIST 1.951,.01= TI(17) to O(48,-1,2,0,0,1)
DIST 2.104,.01= TI(17) to O(53)
DIST 2.044,.01= TI(17) to O(54)
DIST 2.135,.01= TI(17) to O(55)
DIST 1.888,.01= FE(18) to O(46,-1,1,0,0,1)
DIST 1.951,.01= FE(18) to O(48,-1,2,0,-1,1)
DIST 1.951,.01= FE(18) to O(48,-1,2,0,0,1)
DIST 2.104,.01= FE(18) to O(53)
DIST 2.044,.01= FE(18) to O(54)
DIST 2.135,.01= FE(18) to O(55)
DIST 1.950,.01= TI(19) to O(45,-1,2,0,-1,1)
DIST 1.950,.01= TI(19) to O(45,-1,2,0,0,1)
DIST 1.891,.01= TI(19) to O(55)
DIST 1.908,.01= TI(19) to O(56)
DIST 2.104,.01= TI(19) to O(58)
DIST 2.123,.01= TI(19) to O(59)
DIST 1.950,.01= FE(20) to O(45,-1,2,0,-1,1)
DIST 1.950,.01= FE(20) to O(45,-1,2,0,0,1)
DIST 1.891,.01= FE(20) to O(55)
DIST 1.908,.01= FE(20) to O(56)
DIST 2.104,.01= FE(20) to O(58)
DIST 2.123,.01= FE(20) to O(59)
DIST 2.069,.01= TI(21) to O(45,-1,1,0,0,1)
DIST 2.135,.01= TI(21) to O(46,-1,1,0,0,1)
DIST 1.888,.01= TI(21) to O(55)
DIST 1.943,.01= TI(21) to O(56)
DIST 1.951,.01= TI(21) to O(58,1,2,-1,-1)
DIST 1.951,.01= TI(21) to O(58,1,2,-1)
DIST 2.069,.01= FE(22) to O(45,-1,1,0,0,1)
DIST 2.135,.01= FE(22) to O(46,-1,1,0,0,1)
DIST 1.888,.01= FE(22) to O(55)
DIST 1.943,.01= FE(22) to O(56)
DIST 1.951,.01= FE(22) to O(58,1,2,-1,-1)
DIST 1.951,.01= FE(22) to O(58,1,2,-1)
DIST 1.952,.01= TI(23) to O(44,-1,2,0,-1,1)
DIST 1.952,.01= TI(23) to O(44,-1,2,0,0,1)
DIST 1.900,.01= TI(23) to O(57)
DIST 2.131,.01= TI(23) to O(59)
DIST 2.327,.01= TI(23) to O(60)
DIST 1.985,.01= TI(23) to O(61)
DIST 1.952,.01= FE(24) to O(44,-1,2,0,-1,1)
DIST 1.952,.01= FE(24) to O(44,-1,2,0,0,1)
DIST 1.900,.01= FE(24) to O(57)
DIST 2.131,.01= FE(24) to O(59)
DIST 2.327,.01= FE(24) to O(60)
DIST 1.985,.01= FE(24) to O(61)
DIST 1.944,.01= TI(25) to O(43,-1,1,0,0,1)
DIST 2.258,.01= TI(25) to O(44,-1,1,0,0,1)
DIST 2.020,.01= TI(25) to O(57)
DIST 1.907,.01= TI(25) to O(60,1,2,-1,-1)
DIST 1.907,.01= TI(25) to O(60,1,2,-1)
DIST 1.838,.01= TI(25) to O(61)
DIST 1.944,.01= FE(26) to O(43,-1,1,0,0,1)
DIST 2.258,.01= FE(26) to O(44,-1,1,0,0,1)
DIST 2.020,.01= FE(26) to O(57)
DIST 1.907,.01= FE(26) to O(60,1,2,-1,-1)
DIST 1.907,.01= FE(26) to O(60,1,2,-1)
DIST 1.838,.01= FE(26) to O(61)
DIST 1.952,.01= TI(27) to O(42,-1,2,0,-1,1)
DIST 1.952,.01= TI(27) to O(42,-1,2,0,0,1)
DIST 1.915,.01= TI(27) to O(43,-1,1,0,0,1)
DIST 2.038,.01= TI(27) to O(61)
DIST 2.118,.01= TI(27) to O(62)
DIST 1.985,.01= TI(27) to O(63)
DIST 1.952,.01= FE(28) to O(42,-1,2,0,-1,1)
DIST 1.952,.01= FE(28) to O(42,-1,2,0,0,1)
DIST 1.915,.01= FE(28) to O(43,-1,1,0,0,1)
DIST 2.038,.01= FE(28) to O(61)
DIST 2.118,.01= FE(28) to O(62)
DIST 1.985,.01= FE(28) to O(63)
DIST 1.854,.01= TI(29) to O(37,-1,1,0,0,1)
DIST 1.840,.01= TI(29) to O(38,-1,1,0,0,1)
DIST 1.952,.01= TI(29) to O(41,-1,2,0,-1,1)
DIST 1.952,.01= TI(29) to O(41,-1,2,0,0,1)
DIST 2.131,.01= TI(29) to O(63)
DIST 2.213,.01= TI(29) to O(64)
DIST 1.854,.01= FE(30) to O(37,-1,1,0,0,1)
DIST 1.840,.01= FE(30) to O(38,-1,1,0,0,1)
DIST 1.952,.01= FE(30) to O(41,-1,2,0,-1,1)
DIST 1.952,.01= FE(30) to O(41,-1,2,0,0,1)
DIST 2.131,.01= FE(30) to O(63)
DIST 2.213,.01= FE(30) to O(64)
DIST 2.063,.01= TI(31) to O(36,-1,1,0,0,1)
DIST 2.066,.01= TI(31) to O(37,-1,1,0,0,1)
DIST 2.015,.01= TI(31) to O(40,-1,2,0,-1,1)
DIST 2.015,.01= TI(31) to O(40,-1,2,0,0,1)
DIST 2.235,.01= TI(31) to O(64)
DIST 2.319,.01= TI(31) to O(65)
DIST 2.063,.01= FE(32) to O(36,-1,1,0,0,1)
DIST 2.066,.01= FE(32) to O(37,-1,1,0,0,1)
DIST 2.015,.01= FE(32) to O(40,-1,2,0,-1,1)
DIST 2.015,.01= FE(32) to O(40,-1,2,0,0,1)
DIST 2.235,.01= FE(32) to O(64)
DIST 2.319,.01= FE(32) to O(65)
DIST 2.062,.01= TI(33) to O(35,1,1,0,0,1)
DIST 1.955,.01= TI(33) to O(35,-1,1,0,0,1)
DIST 1.966,.01= TI(33) to O(36,-1,1,0,0,1)
DIST 1.905,.01= TI(33) to O(39,-1,2,0,-1,1)
DIST 1.905,.01= TI(33) to O(39,-1,2,0,0,1)
DIST 2.036,.01= TI(33) to O(65)
DIST 2.062,.01= FE(34) to O(35,1,1,0,0,1)
DIST 1.955,.01= FE(34) to O(35,-1,1,0,0,1)
DIST 1.966,.01= FE(34) to O(36,-1,1,0,0,1)
DIST 1.905,.01= FE(34) to O(39,-1,2,0,-1,1)
DIST 1.905,.01= FE(34) to O(39,-1,2,0,0,1)
DIST 2.036,.01= FE(34) to O(65)
# Remove space after hash to activate next line
# USE LAST
(Again: be wary that you may have to play with the "R" - Set the distance search range as part of the bond-length
restraints menu in GSAS. In this case, setting r=1.5 will get GSAS finding all the bonds correctly. Make sure you check the results and that everything is working as required)
I 2.018000 0.010000 1 35
Y
I 1.822000 0.010000 1 36
Y
I 2.222000 0.010000 1 39
Y
I 2.070000 0.010000 1 40
Y
I 1.929000 0.010000 1 65
Y
Y
I 2.018000 0.010000 2 35
Y
I 1.822000 0.010000 2 36
Y
I 2.222000 0.010000 2 39
Y
I 2.070000 0.010000 2 40
Y
I 1.929000 0.010000 2 65
Y
Y
I 1.900000 0.010000 3 37
Y
I 2.007000 0.010000 3 38
Y
I 2.095000 0.010000 3 40
Y
I 2.159000 0.010000 3 41
Y
I 1.925000 0.010000 3 64
Y
Y
I 1.900000 0.010000 4 37
Y
I 2.007000 0.010000 4 38
Y
I 2.095000 0.010000 4 40
Y
I 2.159000 0.010000 4 41
Y
I 1.925000 0.010000 4 64
Y
Y
I 2.038000 0.010000 5 38
Y
I 2.150000 0.010000 5 42
Y
I 1.985000 0.010000 5 43
Y
I 1.950000 0.010000 5 62
Y
Y
I 1.915000 0.010000 5 63
Y
I 2.038000 0.010000 6 38
Y
I 2.150000 0.010000 6 42
Y
I 1.985000 0.010000 6 43
Y
I 1.950000 0.010000 6 62
Y
Y
I 1.915000 0.010000 6 63
Y
I 2.209000 0.010000 7 44
Y
I 2.270000 0.010000 7 45
Y
I 1.911000 0.010000 7 56
Y
I 2.068000 0.010000 7 57
Y
I 2.054000 0.010000 7 59
Y
Y
I 2.209000 0.010000 8 44
Y
I 2.270000 0.010000 8 45
Y
I 1.911000 0.010000 8 56
Y
I 2.068000 0.010000 8 57
Y
I 2.054000 0.010000 8 59
Y
Y
I 1.991000 0.010000 9 46
Y
I 2.061000 0.010000 9 48
Y
I 2.099000 0.010000 9 49
Y
I 1.925000 0.010000 9 53
Y
Y
I 2.032000 0.010000 9 54
Y
I 1.991000 0.010000 10 46
Y
I 2.061000 0.010000 10 48
Y
I 2.099000 0.010000 10 49
Y
I 1.925000 0.010000 10 53
Y
Y
I 2.032000 0.010000 10 54
Y
I 1.900000 0.010000 11 47
Y
I 2.038000 0.010000 11 49
Y
I 2.171000 0.010000 11 50
Y
I 2.030000 0.010000 11 51
Y
I 1.965000 0.010000 11 52
Y
Y
I 1.900000 0.010000 12 47
Y
I 2.038000 0.010000 12 49
Y
I 2.171000 0.010000 12 50
Y
I 2.030000 0.010000 12 51
Y
I 1.965000 0.010000 12 52
Y
Y
I 1.833000 0.010000 13 47
Y
I 1.950000 0.010000 13 50
Y
Y
I 2.095000 0.010000 13 51
Y
Y
I 2.128000 0.010000 13 52
Y
I 1.833000 0.010000 14 47
Y
I 1.950000 0.010000 14 50
Y
Y
I 2.095000 0.010000 14 51
Y
Y
I 2.128000 0.010000 14 52
Y
I 2.033000 0.010000 15 47
Y
I 2.122000 0.010000 15 49
Y
Y
I 2.262000 0.010000 15 52
Y
I 2.297000 0.010000 15 53
Y
I 1.825000 0.010000 15 54
Y
I 2.033000 0.010000 16 47
Y
I 2.122000 0.010000 16 49
Y
Y
I 2.262000 0.010000 16 52
Y
I 2.297000 0.010000 16 53
Y
I 1.825000 0.010000 16 54
Y
I 1.888000 0.010000 17 46
Y
I 1.951000 0.010000 17 48
Y
Y
I 2.104000 0.010000 17 53
Y
I 2.044000 0.010000 17 54
Y
I 2.135000 0.010000 17 55
Y
I 1.888000 0.010000 18 46
Y
I 1.951000 0.010000 18 48
Y
Y
I 2.104000 0.010000 18 53
Y
I 2.044000 0.010000 18 54
Y
I 2.135000 0.010000 18 55
Y
I 1.950000 0.010000 19 45
Y
Y
I 1.891000 0.010000 19 55
Y
I 1.908000 0.010000 19 56
Y
I 2.104000 0.010000 19 58
Y
I 2.123000 0.010000 19 59
Y
I 1.950000 0.010000 20 45
Y
Y
I 1.891000 0.010000 20 55
Y
I 1.908000 0.010000 20 56
Y
I 2.104000 0.010000 20 58
Y
I 2.123000 0.010000 20 59
Y
I 2.069000 0.010000 21 45
Y
I 2.135000 0.010000 21 46
Y
I 1.888000 0.010000 21 55
Y
I 1.943000 0.010000 21 56
Y
I 1.951000 0.010000 21 58
Y
Y
I 2.069000 0.010000 22 45
Y
I 2.135000 0.010000 22 46
Y
I 1.888000 0.010000 22 55
Y
I 1.943000 0.010000 22 56
Y
I 1.951000 0.010000 22 58
Y
Y
I 1.952000 0.010000 23 44
Y
Y
I 1.900000 0.010000 23 57
Y
I 2.131000 0.010000 23 59
Y
I 2.327000 0.010000 23 60
Y
I 1.985000 0.010000 23 61
Y
I 1.952000 0.010000 24 44
Y
Y
I 1.900000 0.010000 24 57
Y
I 2.131000 0.010000 24 59
Y
I 2.327000 0.010000 24 60
Y
I 1.985000 0.010000 24 61
Y
I 1.944000 0.010000 25 43
Y
I 2.258000 0.010000 25 44
Y
I 2.020000 0.010000 25 57
Y
I 1.907000 0.010000 25 60
Y
Y
I 1.838000 0.010000 25 61
Y
I 1.944000 0.010000 26 43
Y
I 2.258000 0.010000 26 44
Y
I 2.020000 0.010000 26 57
Y
I 1.907000 0.010000 26 60
Y
Y
I 1.838000 0.010000 26 61
Y
I 1.952000 0.010000 27 42
Y
Y
I 1.915000 0.010000 27 43
Y
I 2.038000 0.010000 27 61
Y
I 2.118000 0.010000 27 62
Y
I 1.985000 0.010000 27 63
Y
I 1.952000 0.010000 28 42
Y
Y
I 1.915000 0.010000 28 43
Y
I 2.038000 0.010000 28 61
Y
I 2.118000 0.010000 28 62
Y
I 1.985000 0.010000 28 63
Y
I 1.854000 0.010000 29 37
Y
I 1.840000 0.010000 29 38
Y
I 1.952000 0.010000 29 41
Y
Y
I 2.131000 0.010000 29 63
Y
I 2.213000 0.010000 29 64
Y
I 1.854000 0.010000 30 37
Y
I 1.840000 0.010000 30 38
Y
I 1.952000 0.010000 30 41
Y
Y
I 2.131000 0.010000 30 63
Y
I 2.213000 0.010000 30 64
Y
I 2.063000 0.010000 31 36
Y
I 2.066000 0.010000 31 37
Y
I 2.015000 0.010000 31 40
Y
Y
I 2.235000 0.010000 31 64
Y
I 2.319000 0.010000 31 65
Y
I 2.063000 0.010000 32 36
Y
I 2.066000 0.010000 32 37
Y
I 2.015000 0.010000 32 40
Y
Y
I 2.235000 0.010000 32 64
Y
I 2.319000 0.010000 32 65
Y
I 2.062000 0.010000 33 35
Y
Y
I 1.966000 0.010000 33 36
Y
I 1.905000 0.010000 33 39
Y
Y
I 2.036000 0.010000 33 65
Y
I 2.062000 0.010000 34 35
Y
Y
I 1.966000 0.010000 34 36
Y
I 1.905000 0.010000 34 39
Y
Y
I 2.036000 0.010000 34 65
Y