|
Run Crystals, select File, New and go to the directory where the
HKL data and Dirdif solution is located.
|
|
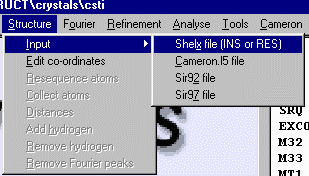
Select Structure, Input, Shelx File (INS or RES) to import the Dirdif solution
which is in a Shelx format. Crystals will prompt for the space group [C 2/c] (which is not part of the
Shelx format), then will continue in importing the information from the Shelx file. If Crystals has a
problem with a kosher Shelx file, it could be you inserted the Space Group file in the wrong format.
|
|
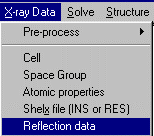
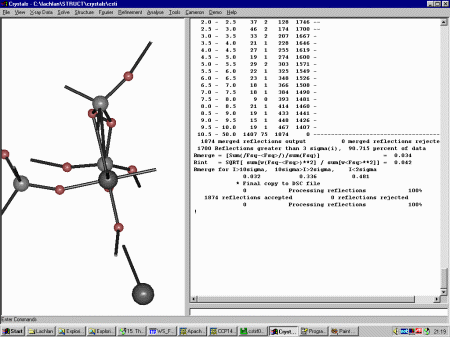
As Crystals can do DLS refinement, reflection data is an option you need to
explicitely set. Thus to insert the Fsquare reflection data, select X-ray Data,
Reflection Data to import the HKL Intensity file.
|
|
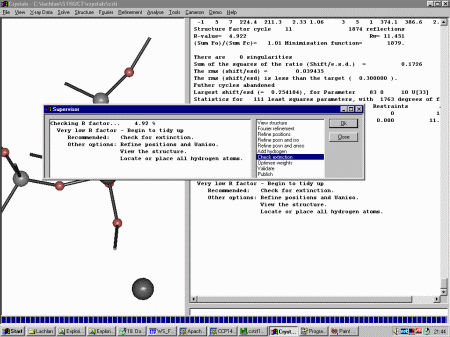
Select Refinement, Guided Automatic to start the refinement. Crystals
will do a few cycles of refinement, then the Supervisor Dialog will then come up to
guide you through the refinement.
|
|
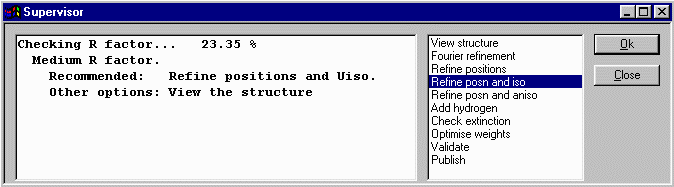
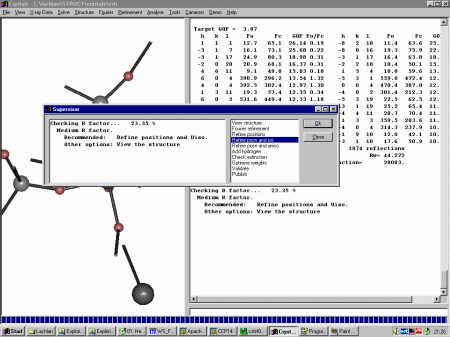
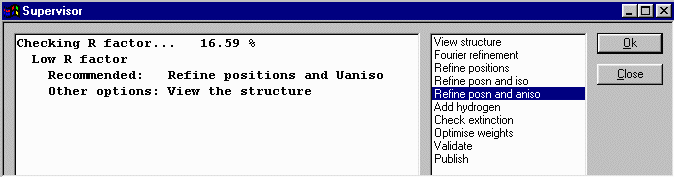
At this point, Crystals recommends we refine position and isotropic thermal
parameters. Click OK to do this. Crystals will then to a cycles of refinement. Crystals now recommends that you continue on with anisotropic refinement but before doing this, it would be best to check out the thermals and other nuances in the Cameron Structure Viewing program as blown up thermals may reveal spurious peaks that were assigned as atoms.
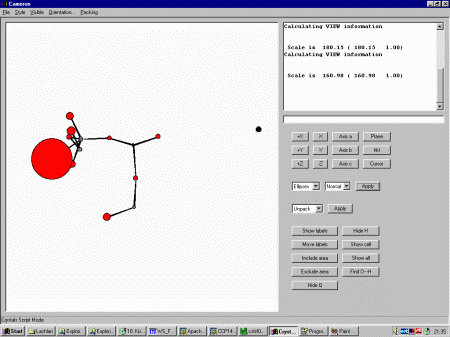
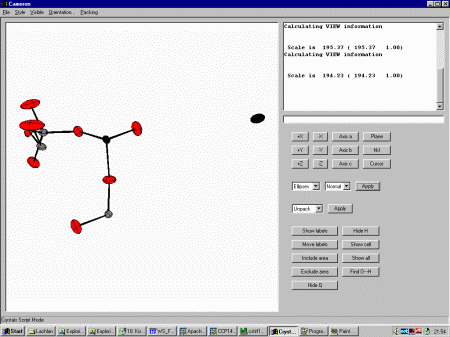
Close the Guided Refinement dialog and select Cameron from
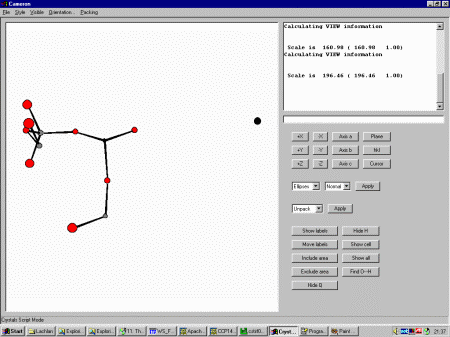
the top menu. Selecting Ellipses shows that one of the Oxygen thermals
has blown up, implying it is a spurious peak that should be deleted.
In the Cameron Command line box (it is still retained for accessing
all the complex and simple functionality of Cameron) type exclude and click
on the offending atom (in this case O14). Then type View to view the result.
Exit Cameron using File, Exit Cameron.
Crystals will prompt whether you wish to apply the changes
performed by Cameron. In this case Yes is the answer.
It should be noted that you can graphically apply commands to
the structure in the Crystals Window (such as deleting, constraints and restraints),
but Cameron is required for closer examination of the structure, looking at packing, etc.
|
|
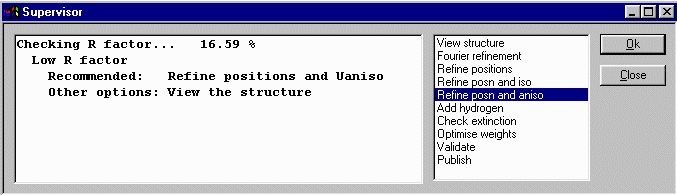
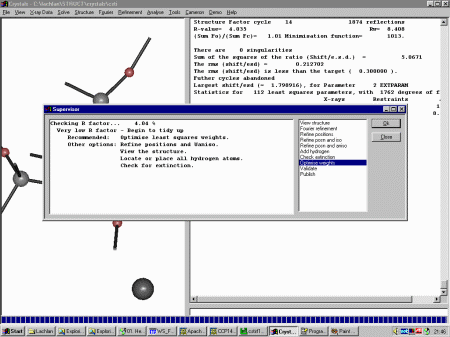
Select Refinement, Guided Automatic to restart the guided refinement.
The Supervisor Dialog will then again come up to guide you through the refinement, in this
case advising you to refine the atomic positions and anisotropic thermals. click OK
to make this happen.
|
|
At this point, Crystals now recommends you Check Extinction. So
continue on with this.
|
|
Crystals now would like to optimise the weighting scheme. Thus continue on
and select "optimal" for weighting scheme determination and go with the defaults.
|
|
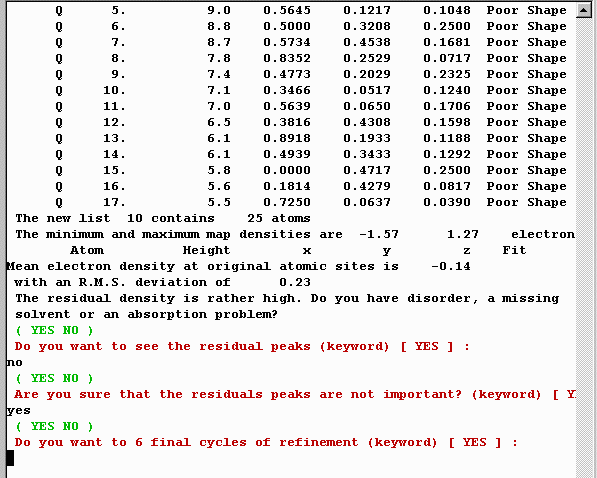
At this point, it may be good to check things out again with
Cameron. The large Oxygen thermals are worth checking out with the
Crystals Fourier Contour Map facility as this may be a split atom problem.
But that will be the subject of another separate tutorial/run-through.
|
|
It is again pointed out that traditional Crystals users and
people who want to issue manual commands can still do this with the bottom
right hand command line dialog box. In this case, we will finish
off the refinement by issuing the manual script command \script routine.
To get out of a script, just type direct at any time.
You can then interact with Crystals in the traditional manner with the added advantage of being able to use the scroll bar to evaluate all the text output.
|
|
CIF files have now been produced and you can exit and restart
Crystals at any time and continue where you left off in the refinement.
|