|
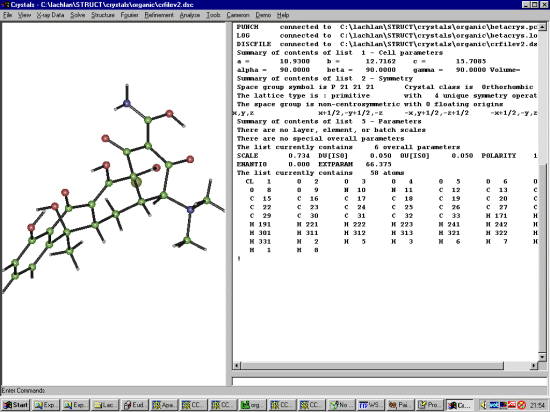
Run Crystals, select File, Open Datafile and go to the directory where the
Structure has been refined in a previous Crystals session. (you can also browse
around via the Windows explorer and double click on the crfilev2.dsc file)
|
|
Use [SHIFT] [LEFT-MOUSE-KEY] to zoom up on the structural
area of interest.
|
|
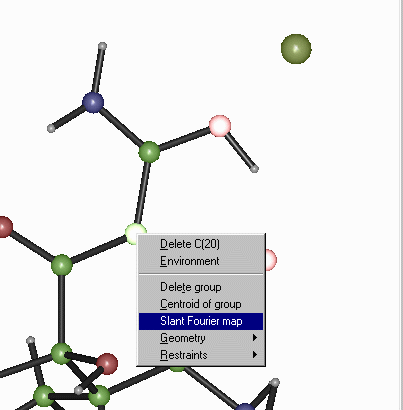
Use the mouse and [LEFT-MOUSE-KEY] to sellect that three atoms that
define the slant plane. When at atom is selected, it will change colour.
|
|
Put the mouse cursor on one of the selected atoms, click [RIGHT-MOUSE-KEY]
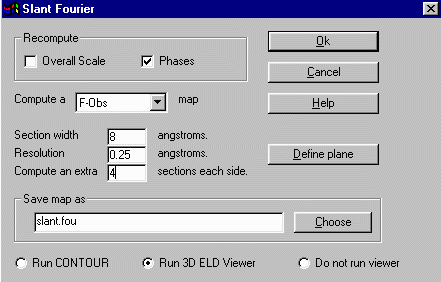
and select Slant Plane Fourier which brings up an options box.
To optimise and get a best view, you might find a "section width" of 8
Angstrom and computing an extra 4 sections each side would be good. Normally
this would probably be determined from experience/trial and error.
Then press OK to continue.
|
|
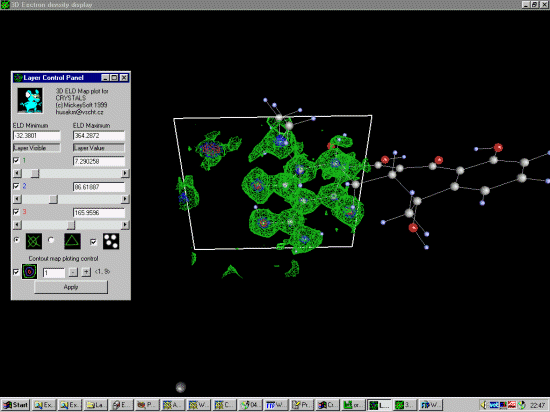
Optimise the 3D contour levels via the slide bars and examine the structure
using the mouse to rotate the structure around.
|